

-臨床成績〈成人〉-
国内第Ⅲ相単盲検比較試験〈成人〉1)
| 目的 |
フルティフォーム®の有効性及び安全性をフルチカゾンプロピオン酸エステル(FP)エアゾール製剤との比較により検討する。 |
|---|---|
| 対象 |
16歳以上の気管支喘息患者455例 選択基準
など |
| 方法 |
無作為化単盲検並行群間比較試験。2週間の観察期間の後、フルティフォーム®群とFP群に割付け、8週間投与した。 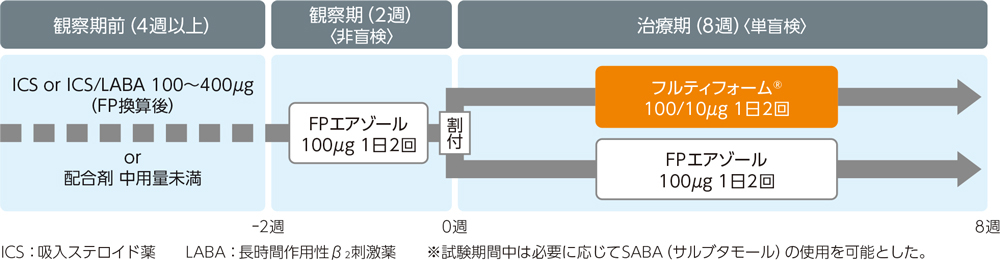
|
| 評価項目 |
有効性評価項目
|
| 解析計画 |
〈主要評価項目〉
〈副次評価項目〉
FEV1:1秒量、FVC:努力肺活量、%PEF:ピークフロー値の基準値に対する測定値の割合、ANCOVA:共分散分析 |
臨床成績
主要評価項目
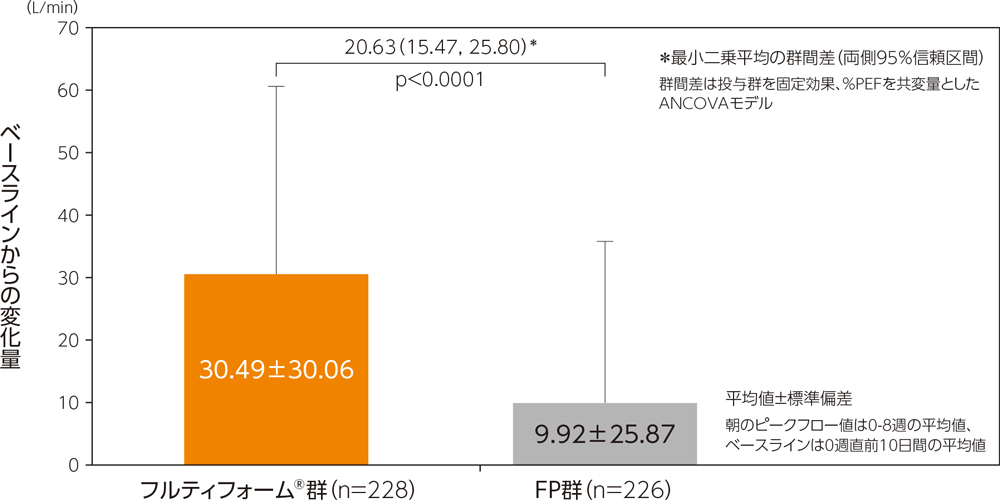
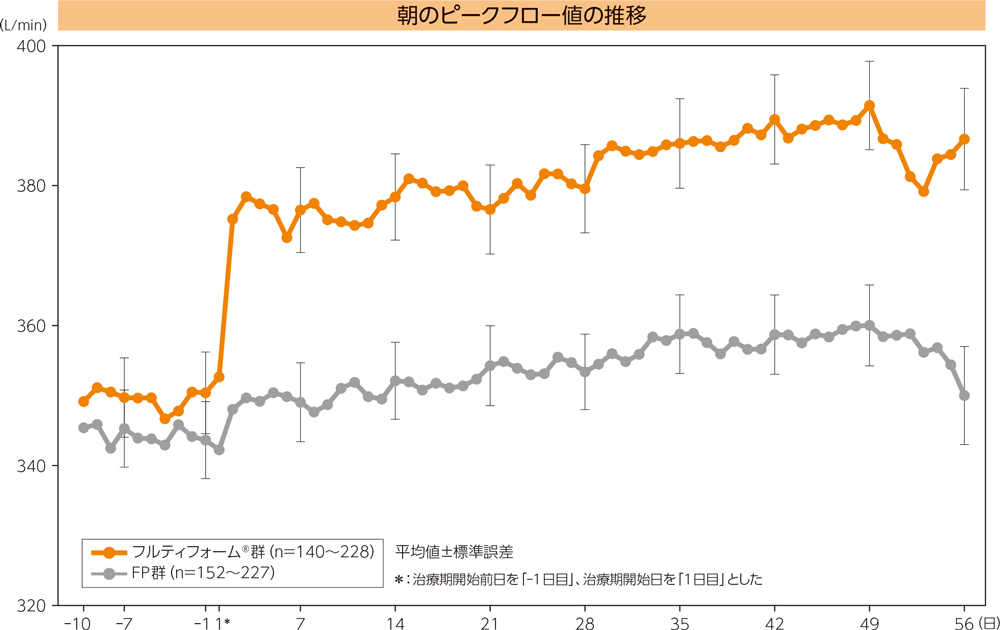
(1)朝のピークフロー値のベースラインからの変化量
朝のピークフロー値(0-8週の平均値)におけるベースラインからの変化量の平均値はフルティフォーム®群で30.49L/min、FP群で9.92L/min、最小二乗平均の群間差(両側95%信頼区間)は20.63L/min(15.47, 25.80)であり、フルティフォーム®群のFP群に対する優越性が検証された(p<0.0001、ANCOVA)。
副次評価項目
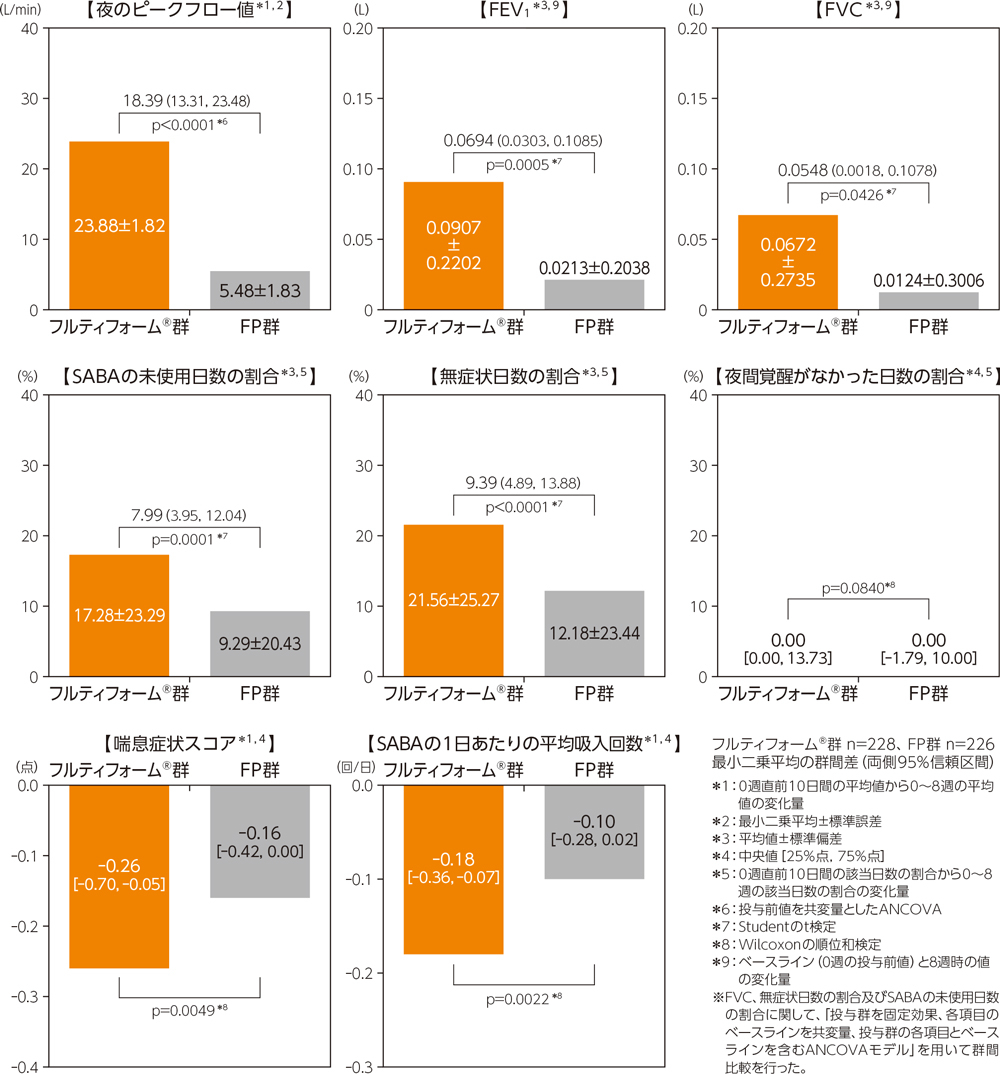
(2)各指標のベースラインからの変化量
喘息症状による夜間覚醒がなかった日数の割合については、両群間で有意差は認められなかったが(Wilcoxonの順位和検定)、夜のピークフロー値(p<0.0001、ANCOVA)、FEV1(p=0.0005、Studentのt検定)、FVC(p=0.0426、Studentのt検定)、SABAの未使用日数の割合(p=0.0001、Studentのt検定)、無症状日数の割合(p<0.0001、Studentのt検定)、喘息症状スコア(p=0.0049、Wilcoxonの順位和検定)、SABAの1日あたりの平均吸入回数(p=0.0022、Wilcoxonの順位和検定)については、フルティフォーム®群ではFP群と比較して有意差が認められた。
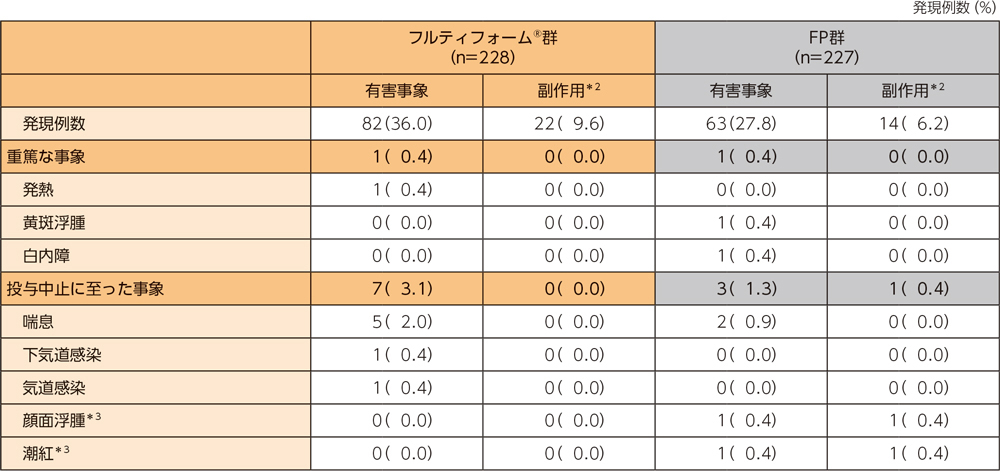
安全性
フルティフォーム®群の有害事象は228例中82例(36.0%)、副作用は228例中22例(9.6%)であった。
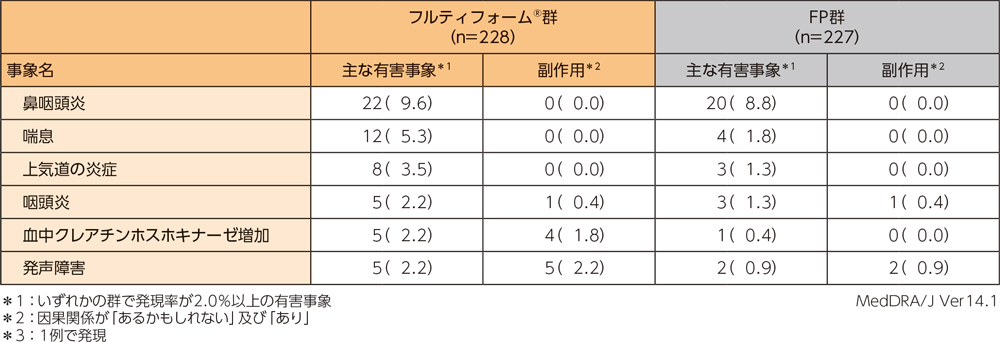
フルティフォーム®群の重篤な有害事象、投与中止に至った有害事象、主な有害事象、主な副作用はそれぞれ以下のとおりである。本試験において、死亡例は認められなかった。
国内第Ⅲ相長期投与試験〈成人〉2)3)
| 目的 |
フルティフォーム®の52週間長期投与による安全性及び有効性を検討する。 |
|---|---|
| 対象 |
16歳以上の気管支喘息患者244例(完了被験者214例) |
| 方法 |
非盲検非対照試験。2週間の観察期間の後、観察期で使用した吸入ステロイド薬(ICS)の用量に基づき、低用量群、中用量群、高用量群に割付け、52週間投与した。また、治療期間中、増量又は減量基準に合致した場合には、増量又は減量を可能とした。 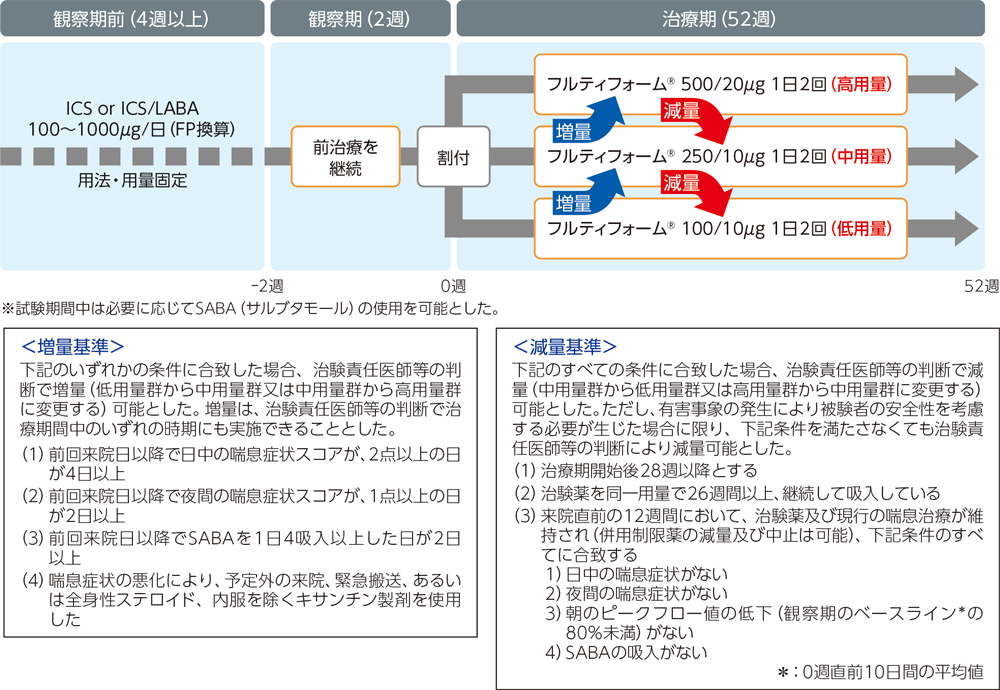
|
| 評価項目 |
|
| 解析計画 |
|
5. 効能又は効果に関連する注意
- 5.1
-
患者、保護者又はそれに代わる適切な者に対し次の注意を与えること。
本剤は発現した発作を速やかに軽減する薬剤ではないので、急性の発作に対しては使用しないこと。
- 5.2
- 本剤の投与開始前には、患者の喘息症状を比較的安定な状態にしておくこと。特に、喘息発作重積状態又は喘息の急激な悪化状態のときには原則として本剤は使用しないこと。
7. 用法及び用量に関連する注意
症状の緩解がみられた場合は、治療上必要最小限の用量を投与し、必要に応じ吸入ステロイド剤への切り替えも考慮すること。
臨床成績
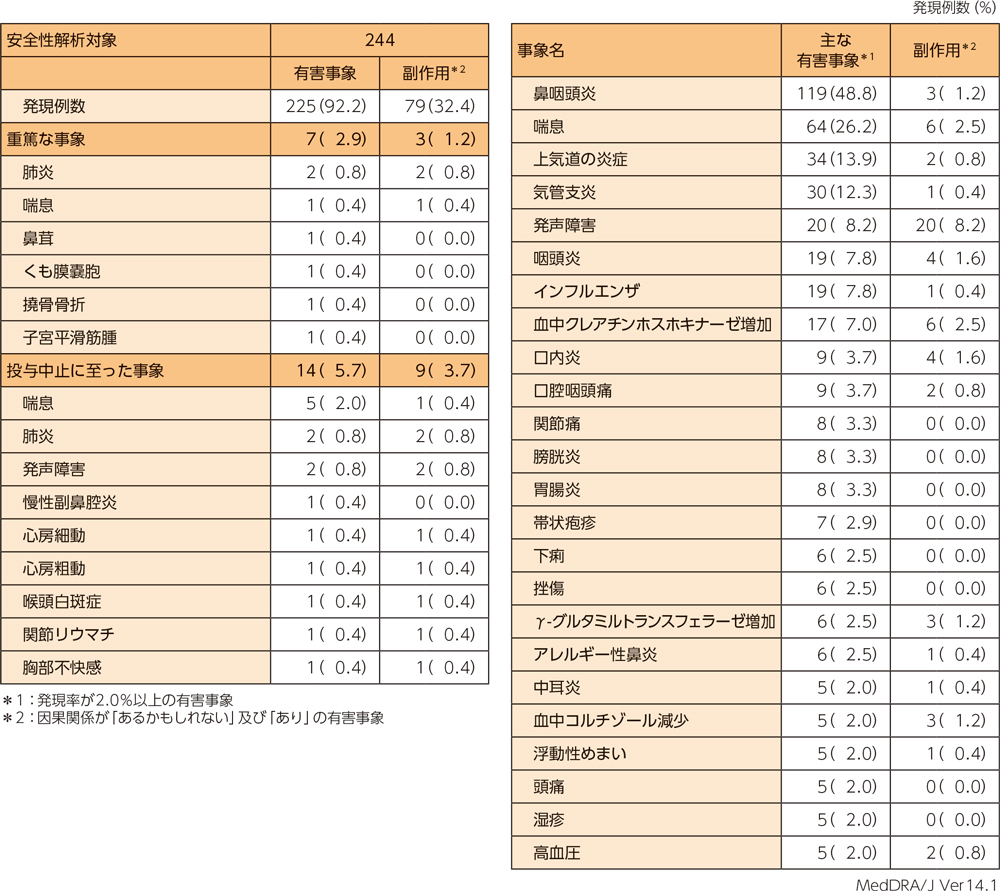
安全性評価項目
(1)有害事象及び副作用
有害事象は244例中225例(92.2%)、副作用は244例中79例(32.4%)に認められた。
重篤な有害事象、中止に至った有害事象、主な有害事象、主な副作用はそれぞれ以下のとおりである。本試験において、死亡例は認められなかった。
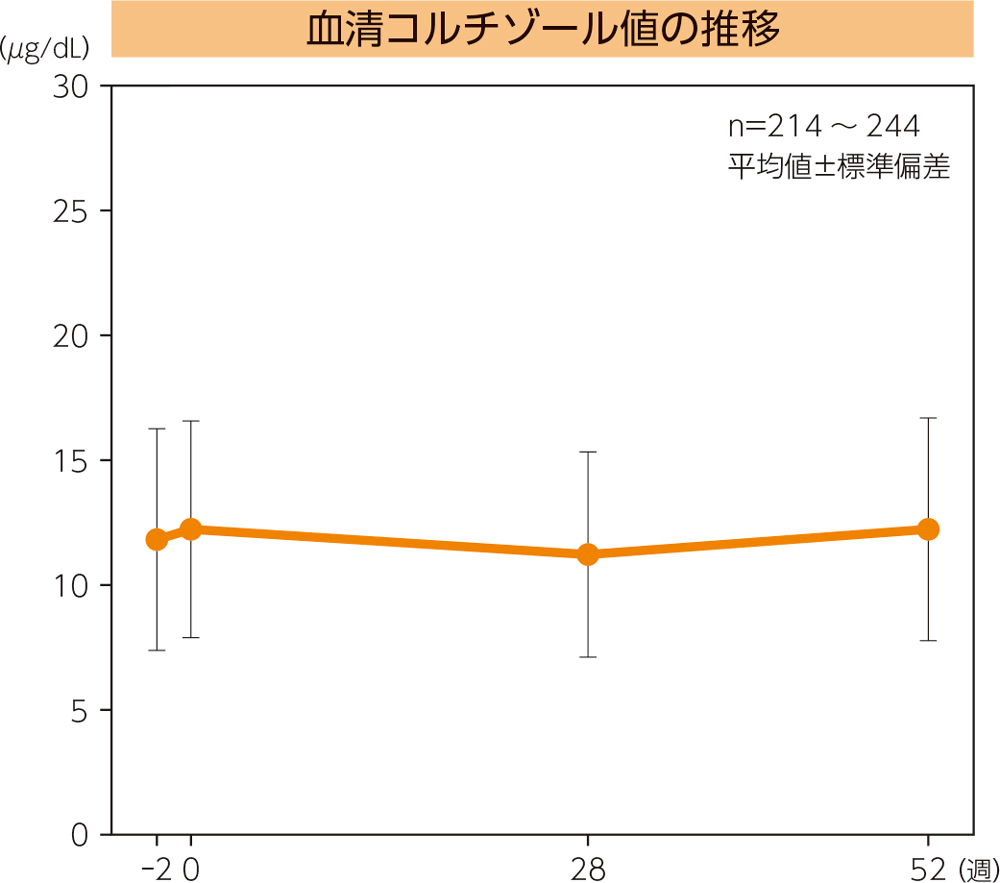
(2)血清コルチゾール値の推移
血清コルチゾール平均値の推移は下図の通りである。なお、試験期間中に血清コルチゾール低下に関する有害事象は6例(血中コルチゾール減少5例、腎機能不全1例)に認められ、このうち4例(100/10μg:1例、500/20μg:3例)が治験薬との因果関係は否定できないと判断された。これら4例の重症度は軽度又は中等度であった。
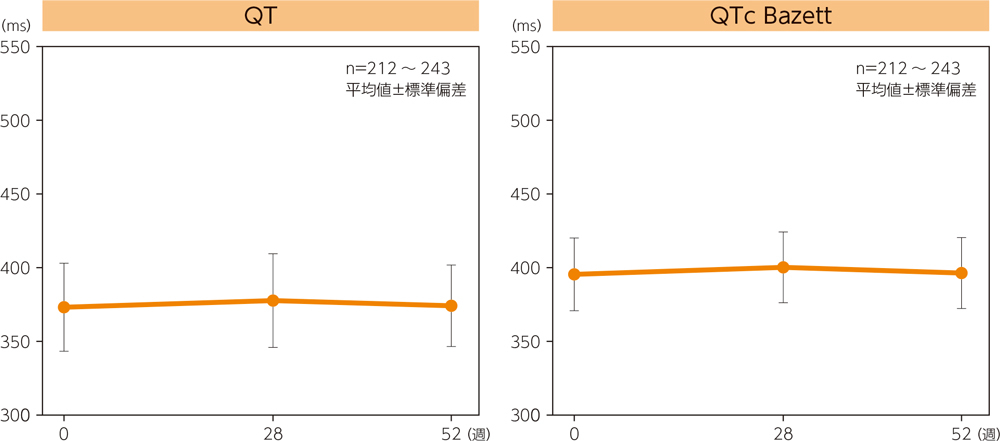
(3)心電図パラメータの推移
心電図パラメータ平均値の推移は下図の通りである。各検査時期を通してQTcの絶対値が500msを超える被験者がQTc(Bazett)で1例(検査時投与量:500/20μg)、0週からの変化量で60msを超える被験者がQTc(Bazett)で5例(250/10μg 3例、500/20μg 2例)、QTc(Fridericia)で1例(250/10μg)認められた。なお、心電図検査における有害事象は6例(心房細動及び心房粗動、心房細動、第一度房室ブロック、左脚ブロック、洞房ブロック、洞性頻脈各1例)に認められ、このうち5例(心房細動及び心房粗動、心房細動、第一度房室ブロック、洞房ブロック、洞性頻脈各1例)は因果関係が否定できない(副作用)と判断された。このうち、1例(心房細動及び心房粗動)は重症度が高度であり投与中止に至った。
有効性(副次評価項目)
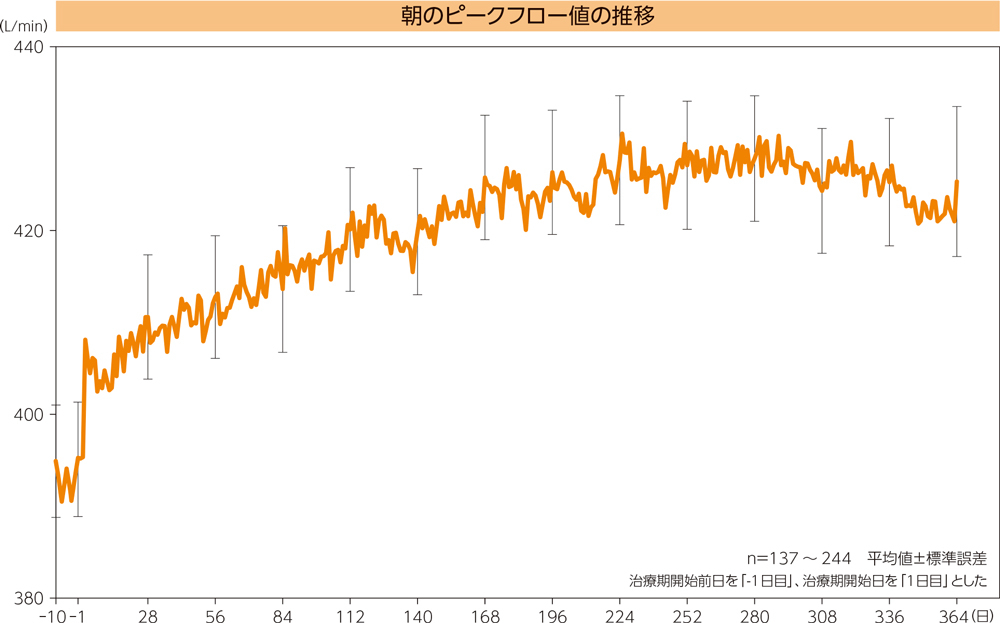
(1)朝のピークフロー値2)
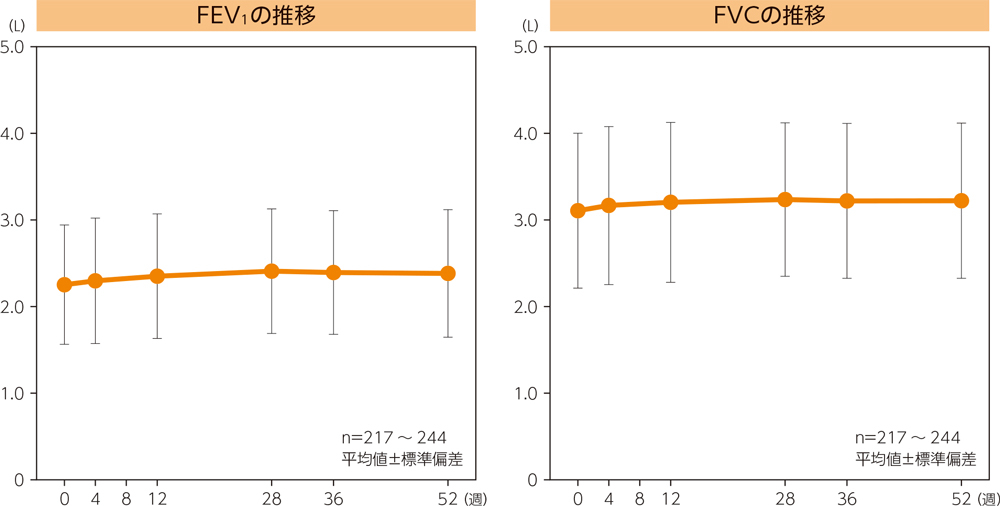
(2)呼吸機能検査(FEV1、FVC値の推移)
(3)その他の評価項目
朝のピークフロー値、夜のピークフロー値、喘息症状スコア、無症状日数の割合、喘息症状による夜間覚醒がなかった日数の割合、SABAの未使用日数の割合、SABAの1日あたりの平均吸入回数は、ベースラインと比較して、投与期間の最終2週間において有意差が認められた(有意水準両側5%、いずれもp<0.0001、Wilcoxonの符号付き順位和検定)。
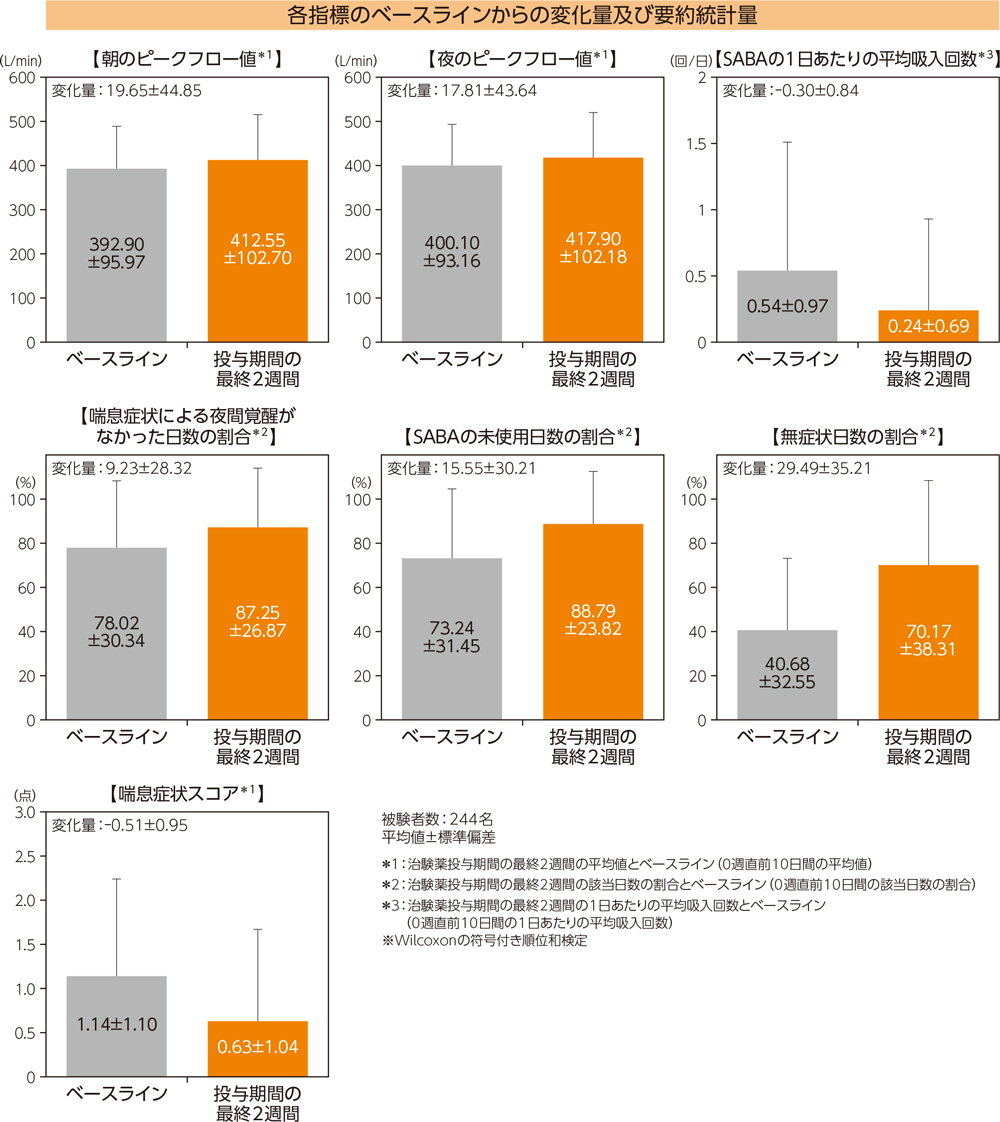
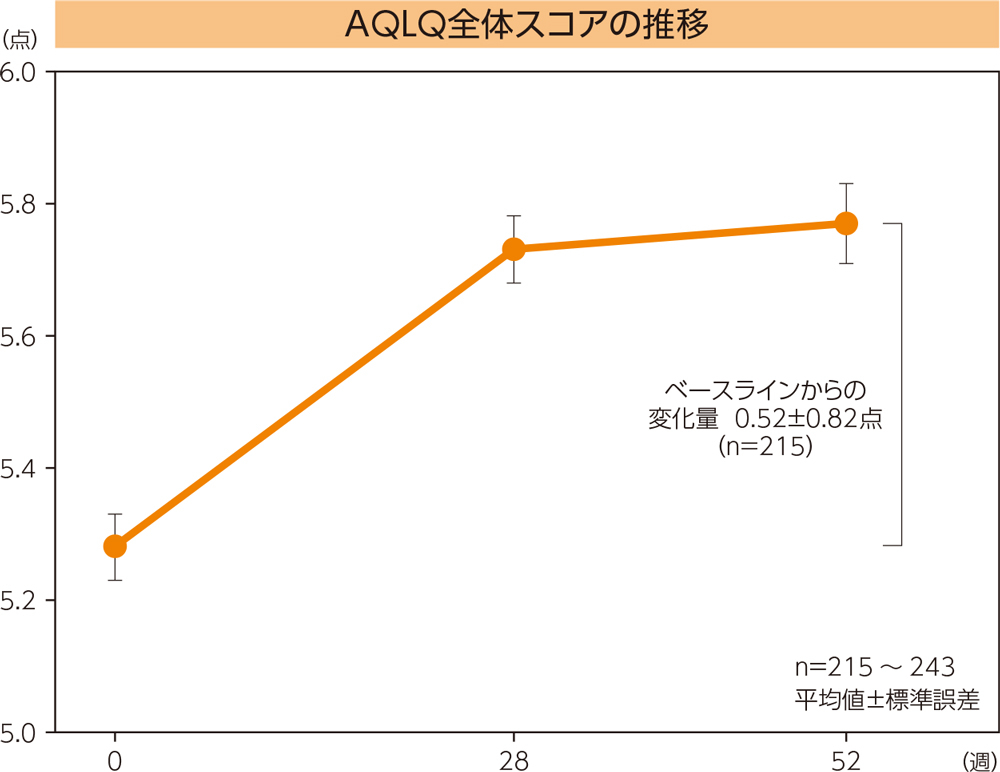
AQLQ
AQLQ全体スコアの52週時におけるベースラインからの変化量は0.52±0.82(平均値±標準偏差)であった。
- 【参考】
-
AQLQ(Asthma Quality of Life
Questionaire:喘息患者のQOLに関する調査票)は、活動制限、感情機能、症状、環境刺激の4項目、合計32の質問で構成されている質問票であり、7点のスケール(7=非常に良い、1=非常に悪い)で評価する。
0.5点以上の増加が臨床的に意義のある改善とされている。
Juniper EF et al. J Clin Epidemiol 47;81, 1994.
5. 効能又は効果に関連する注意
- 5.1
-
患者、保護者又はそれに代わる適切な者に対し次の注意を与えること。
本剤は発現した発作を速やかに軽減する薬剤ではないので、急性の発作に対しては使用しないこと。
- 5.2
- 本剤の投与開始前には、患者の喘息症状を比較的安定な状態にしておくこと。特に、喘息発作重積状態又は喘息の急激な悪化状態のときには原則として本剤は使用しないこと。
7. 用法及び用量に関連する注意
症状の緩解がみられた場合は、治療上必要最小限の用量を投与し、必要に応じ吸入ステロイド剤への切り替えも考慮すること。
- 1)フルティフォーム®の国内第Ⅲ相単盲検比較試験〈成人〉(承認時評価資料).
- 2)フルティフォーム®の国内第Ⅲ相長期投与試験〈成人〉(承認時評価資料).
-
3)東田有智 他. アレルギー・免疫 20; 1686, 2013.
利益相反:本研究は杏林製薬株式会社の支援により行われた。
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。