-国際共同第Ⅲ相試験-
探索的なPPK解析、並びに有効性及び安全性のデータに基づく探索的なER解析の結果から本試験の用法及び用量が設定されました。一部承認外用量の投与群が含まれますが、承認審査過程で評価された試験成績であるため、試験成績を紹介しております。
1. 国際共同第Ⅲ相試験(027試験; COUGH-1)1)2)
| 目的 |
治療抵抗性又は原因不明の慢性咳嗽に対するリフヌア®錠45mg及び15mg 1日2回経口投与の有効性に関しプラセボに対する優越性を検証し、安全性を評価する。 |
|---|---|
| 対象 |
咳嗽が1年以上継続している18歳以上の治療抵抗性又は原因不明の慢性咳嗽患者732例(日本人34例を含む)
[定義]
治療抵抗性又は原因不明の慢性咳嗽の診断は米国胸部疾患学会(ACCP)ガイドライン20063)に基づいた。 |
| 試験デザイン |
多施設共同、無作為化、二重盲検、プラセボ対照、並行群間比較試験(国際共同試験) |
| 方法 |
リフヌア®錠45mg群、リフヌア®錠15mg群又はプラセボ群に1:1:1の比で無作為に割り付けた。 |

|
|
| 評価項目 |
主要評価項目:
副次評価項目:
安全性評価項目:有害事象、副作用など ※咳嗽頻度以外は患者報告アウトカム(PRO)評価。質問票解説とレスポンダーの定義は下記参照 |
| 解析計画 |
|
- FAS(Full Analysis Set):
- 無作為割り付け後に1回以上治験薬の投与を受け、評価項目についてベースライン時と投与期間中の投与後に各1回以上測定したすべての被験者
- APaT(All-Participants-as-Treated):
- 無作為割り付け後に治験薬を1回以上投与されたすべての被験者
- 6.
-
用法及び用量
通常、成人にはゲーファピキサントとして1回45mgを1日2回経口投与する。
患者背景(APaT)
| リフヌア®錠45mg群 (n=243) |
リフヌア®錠15mg群 (n=244) |
プラセボ群 (n=243) |
|
|---|---|---|---|
性別 例数(%) |
|||
| 男性 | 63(25.9) | 63(25.8) | 62(25.5) |
| 女性 | 180(74.1) | 181(74.2) | 181(74.5) |
| 年齢(歳) | |||
| 平均(標準偏差) | 59.4(13.1) | 59.6(11.7) | 57.9(13.1) |
| BMI(kg/m2) | |||
| 平均(標準偏差) | 27.96(5.86) | 28.72(5.80) | 28.04(5.56) |
慢性咳嗽の期間(年) 例数(%) |
|||
| 10年未満 | 134(55.1) | 130(53.3) | 127(52.3) |
| 10年以上 | 109(44.9) | 114(46.7) | 116(47.7) |
| 平均(標準偏差) | 11.24(9.36) | 11.80(9.13) | 11.72(9.92) |
慢性咳嗽の診断
例数(%) | |||
| 治療抵抗性 | 139(57.2) | 141(57.8) | 148(60.9) |
| 原因不明 | 104(42.8) | 103(42.2) | 95(39.1) |
併存疾患 例数(%) |
|||
| あり | 243(100.0) | 244(100.0) | 243(100.0) |
| 喘息 | 106(43.6) | 94(38.5) | 97(39.9) |
| 胃食道逆流性疾患 | 88(36.2) | 100(41.0) | 108(44.4) |
| アレルギー性鼻炎 | 48(19.8) | 48(19.7) | 48(19.8) |
前治療薬 例数(%) |
|||
| あり | 237(97.5) | 235(96.3) | 234(96.3) |
| 閉塞性気道疾患治療薬 | 183(75.3) | 178(73.0) | 182(74.9) |
| 咳嗽及び感冒治療薬 | 99(40.7) | 99(40.6) | 98(40.3) |
| 点鼻薬 | 145(59.7) | 139(57.0) | 147(60.5) |
| 全身性抗ヒスタミン薬 | 96(39.5) | 88(36.1) | 98(40.3) |
| 酸関連疾患治療薬 | 147(60.5) | 143(58.6) | 146(60.1) |
| ベースライン時の平均週間咳重症度VAS(mm) | (n=243) | (n=244) | (n=241) |
| 平均(標準偏差) | 67.88(12.83) | 68.20(15.01) | 69.12(13.85) |
| ベースライン時の24時間の咳嗽頻度(回/時間) | (n=237) | (n=235) | (n=232) |
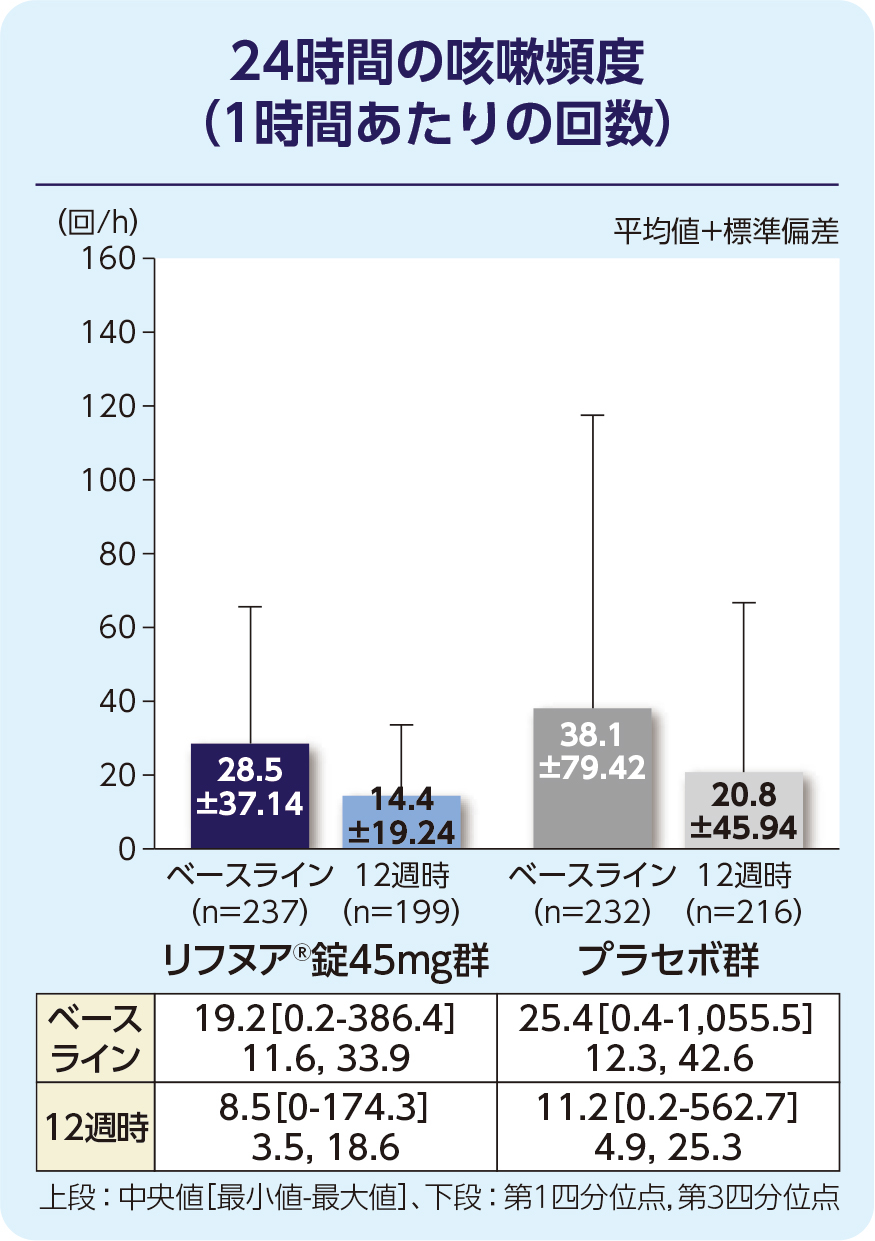
| 平均(標準偏差) | 28.53(37.14) | 26.79(21.13) | 38.07(79.42) |
| ベースライン時のLCQ合計スコア | (n=236) | (n=233) | (n=229) |
| 平均(標準偏差) | 10.5(2.73) | 10.5(2.86) | 10.0(3.08) |
| ベースライン時のCSD合計スコア | (n=243) | (n=244) | (n=241) |
| 平均(標準偏差) | 6.1(1.45) | 6.1(1.69) | 6.2(1.53) |
| 合計HARQスコア | (n=240) | (n=235) | (n=239) |
| 平均(標準偏差) | 39.25(12.99) | 39.37(13.29) | 40.18(13.55) |
14項目(70点満点)からなる慢性咳嗽の主要な要素を抽出することを⽬的とした質問票
- 6.
-
用法及び用量
通常、成人にはゲーファピキサントとして1回45mgを1日2回経口投与する。
リフヌア®錠15mg群は、承認外の用法及び用量のため、有効性の結果からは削除しています。
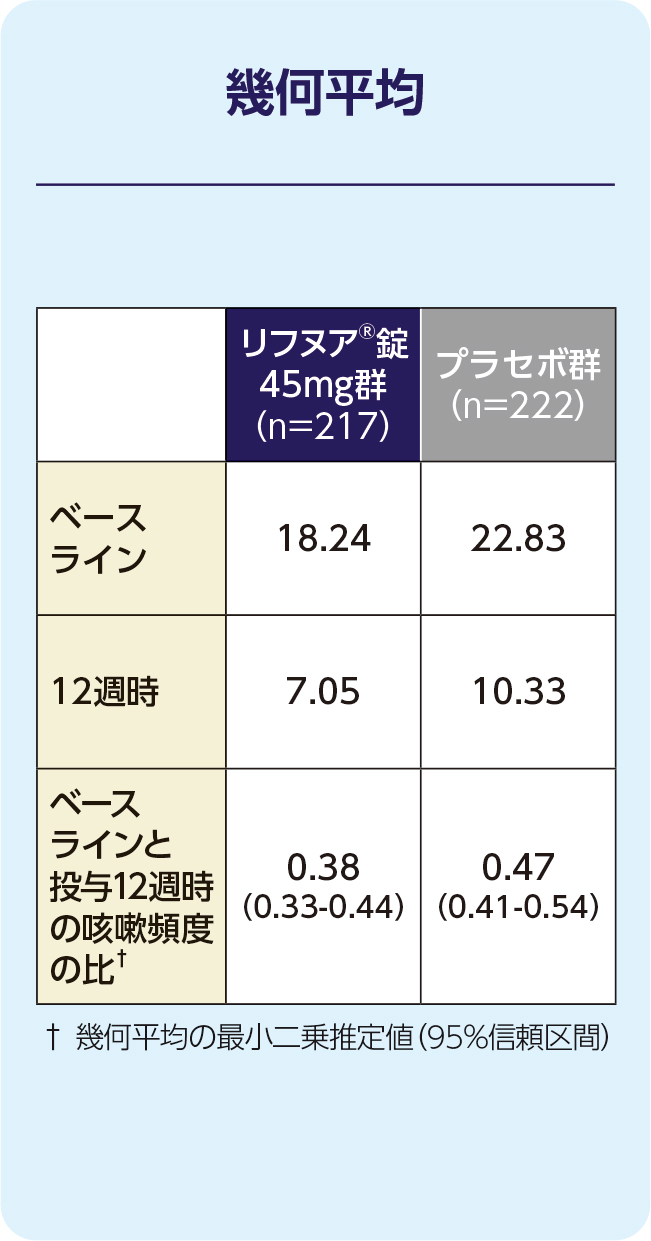
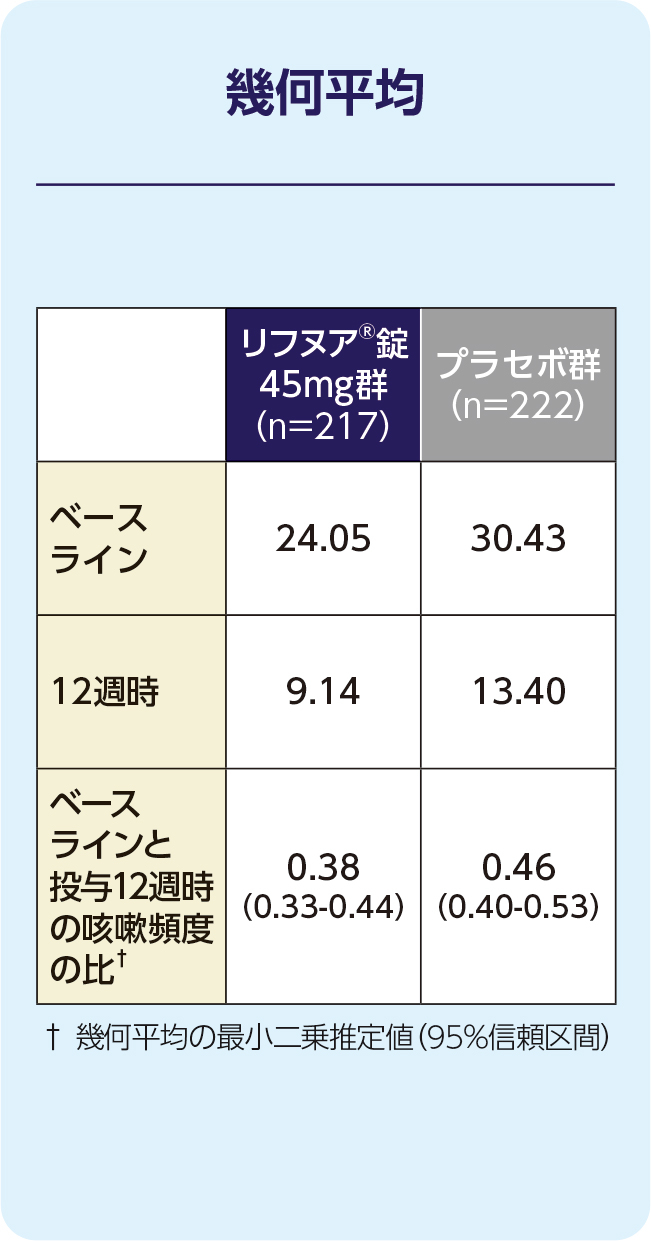
(1)12週時の24時間の咳嗽頻度(1時間あたりの回数)(FAS)[主要評価項目]
12週時の24時間の咳嗽頻度(1時間あたりの回数)の減少に関して、リフヌア®錠45mg群はプラセボ群に対する優越性が検証されました(p=0.041; 経時データ型共分散分析モデル*)。プラセボ群に対する相対減少率は-18.45%(95%信頼区間: -32.92, -0.86)でした。

- *
- 対数変換後の24時間の咳嗽頻度の各時点のベースラインからの変化量に対して、投与群、時点、投与群と時点の交互作用項、性別、地域を固定効果、対数変換後のベースラインの咳嗽頻度、対数変換後のベースラインの咳嗽頻度と時点の交互作用項を共変量とした経時データ型共分散分析モデル。時点間の共分散構造には無構造を仮定した。
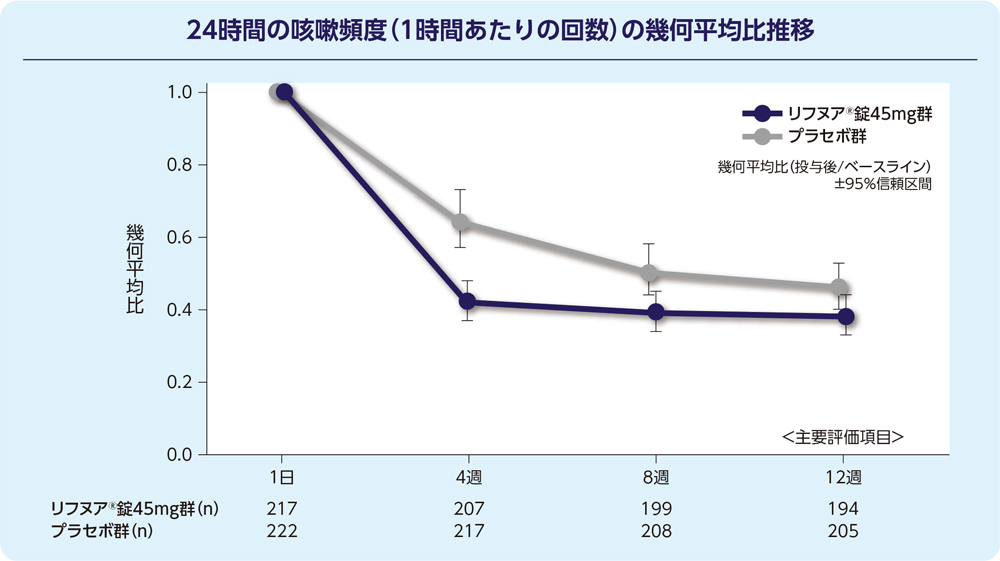
解析計画(事前に規定した固定順序を用いた多重性調整)に基づき、副次評価項目の統計学的検定は実施しなかった。
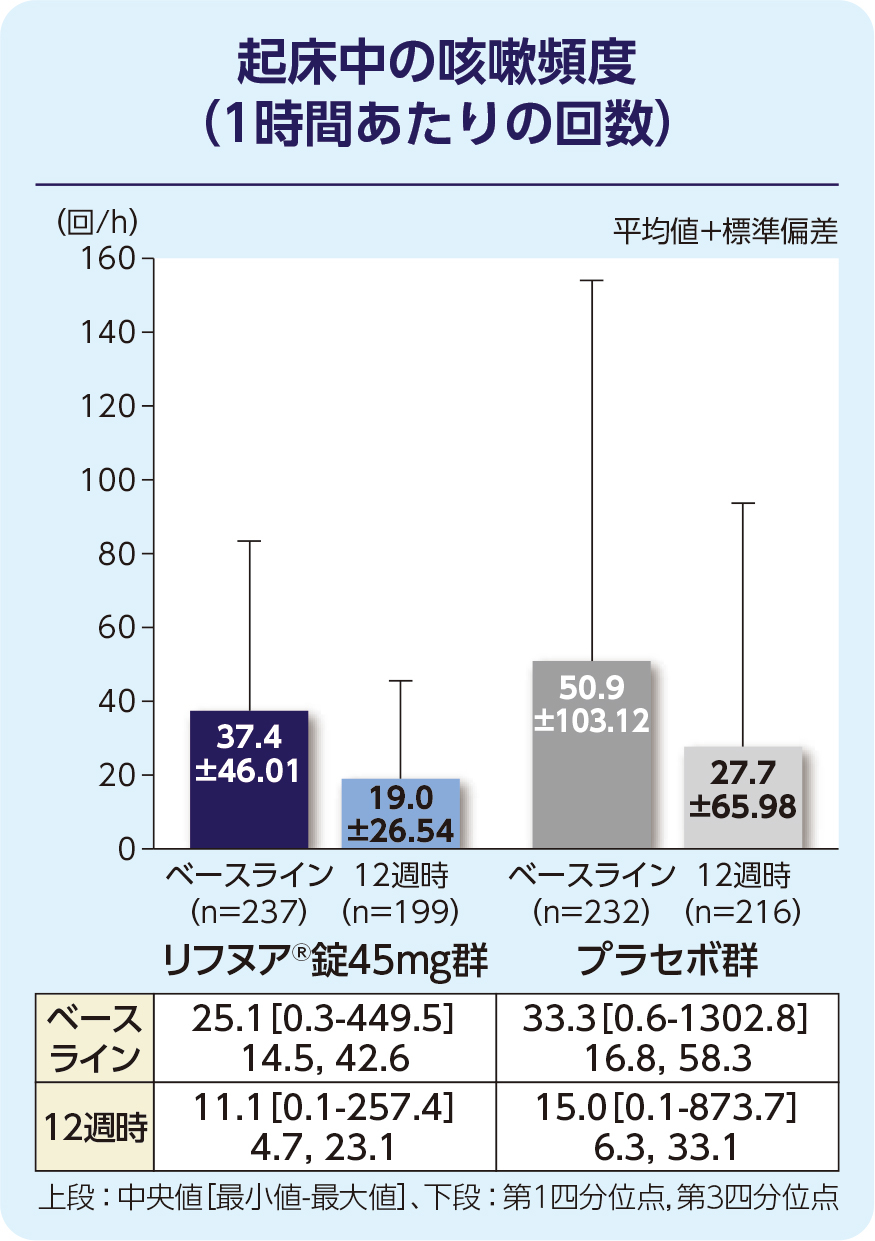
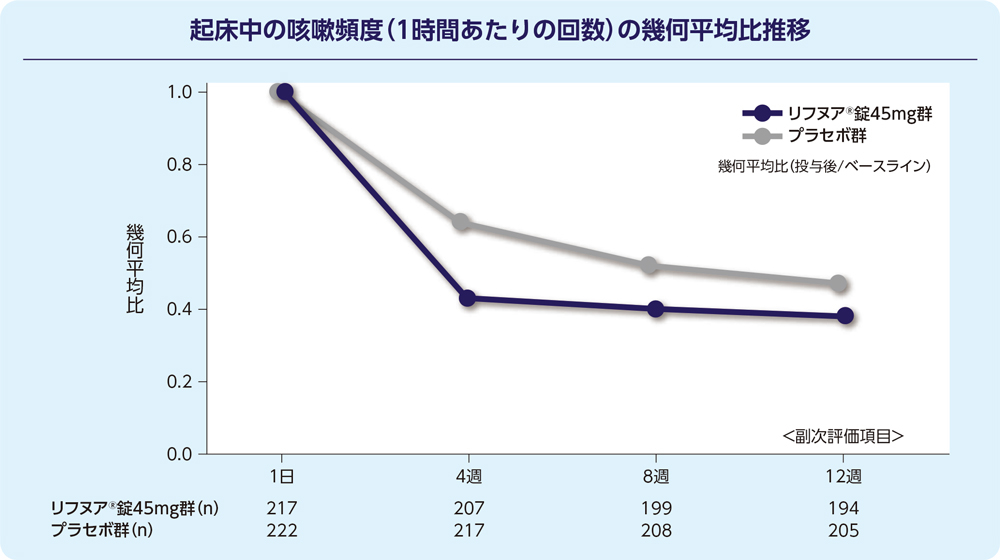
(2)12週時の起床中の咳嗽頻度(1時間あたりの回数)(FAS)[副次評価項目]
12週時の起床中の咳嗽頻度(1時間あたりの回数)のプラセボ群に対する相対減少率は-17.68%でした。

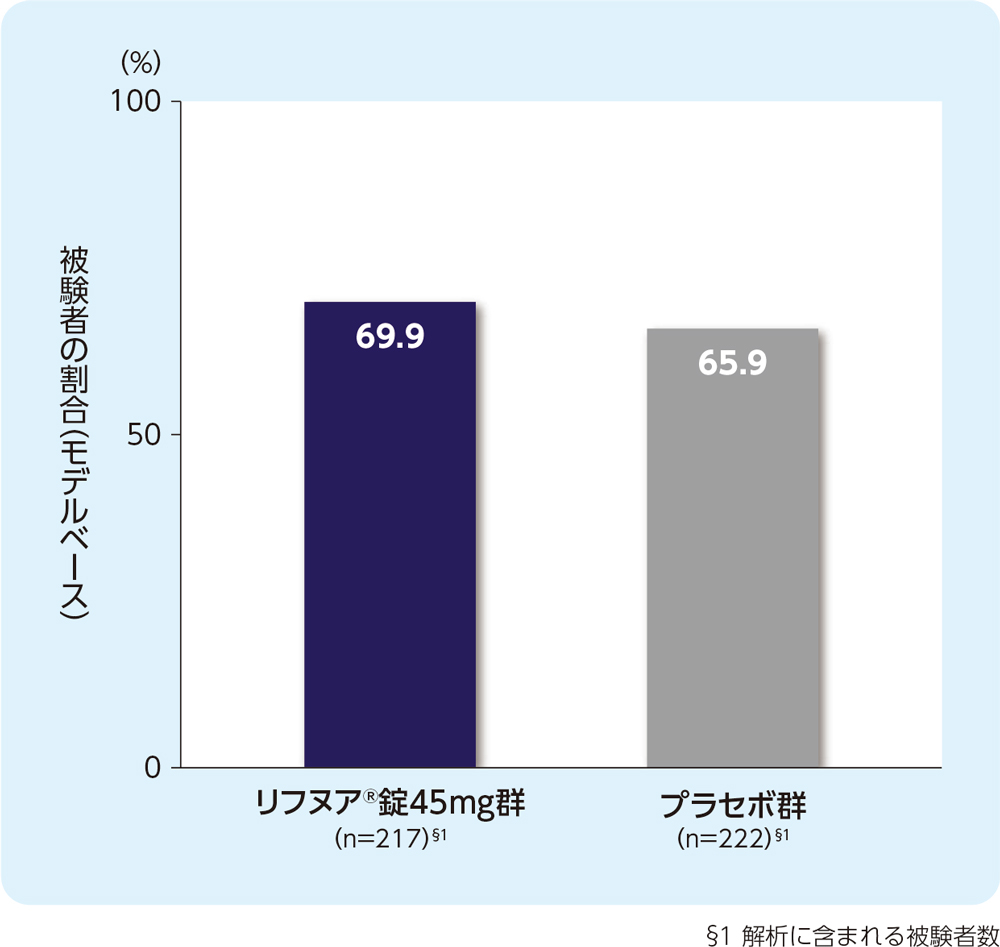
(3) 12週時の24時間の咳嗽頻度(1時間あたりの回数)がベースラインから30%以上減少した被験者の割合(FAS)[副次評価項目]
12週時の24時間の咳嗽頻度(1時間あたりの回数)がベースラインから30%以上減少した被験者の割合は、リフヌア®錠45mg群で69.9%、プラセボ群で65.9%でした。
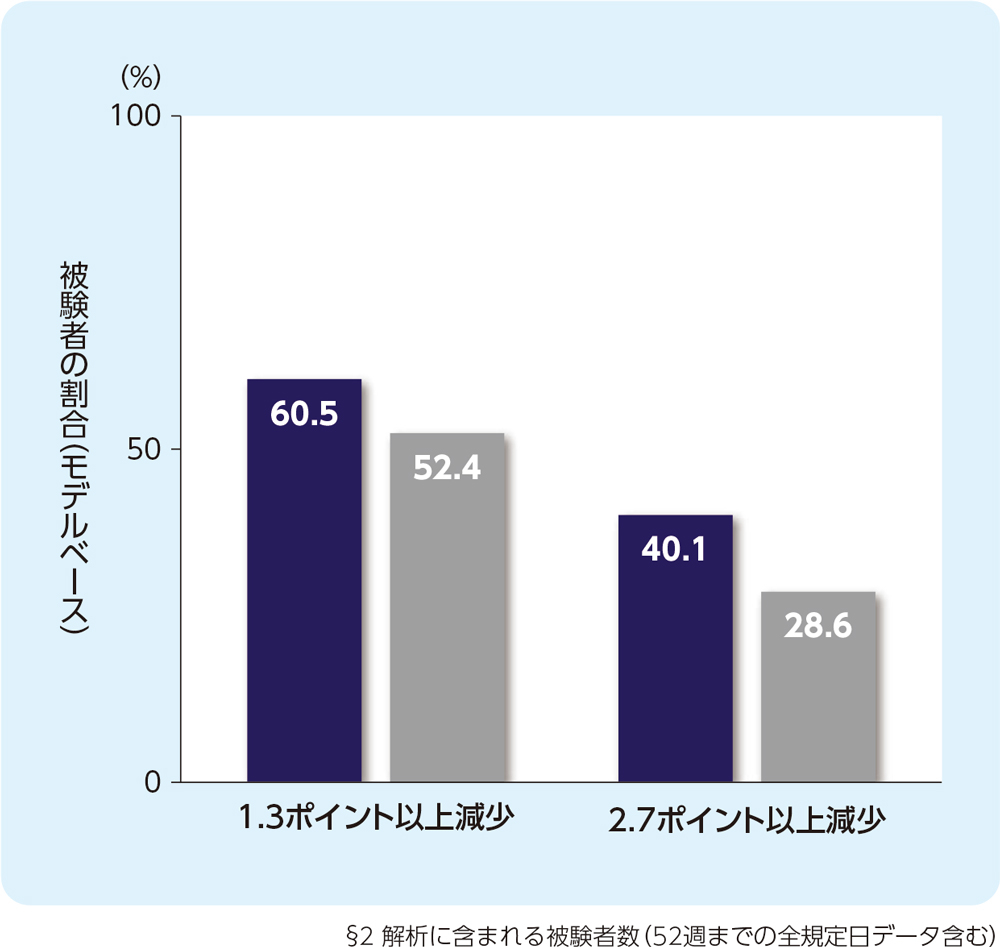
(4)12週時のCSD合計スコアの週平均がベースラインから1.3ポイント以上又は2.7ポイント以上減少した被験者の割合(FAS)[副次評価項目]
12週時のCSD合計スコアの週平均がベースラインから1.3ポイント以上減少した被験者の割合は、リフヌア®錠45mg群で60.5%、プラセボ群で52.4%でした。2.7ポイント以上減少した被験者の割合は、リフヌア®錠45mg群で40.1%、プラセボ群で28.6%でした。
■リフヌア®錠45mg群(n=234)§2
■プラセボ群(n=237)§2
ベースライン値(平均値±標準偏差)
リフヌア®錠45mg群(n=243):6.1±1.45
プラセボ群(n=241):6.2±1.53
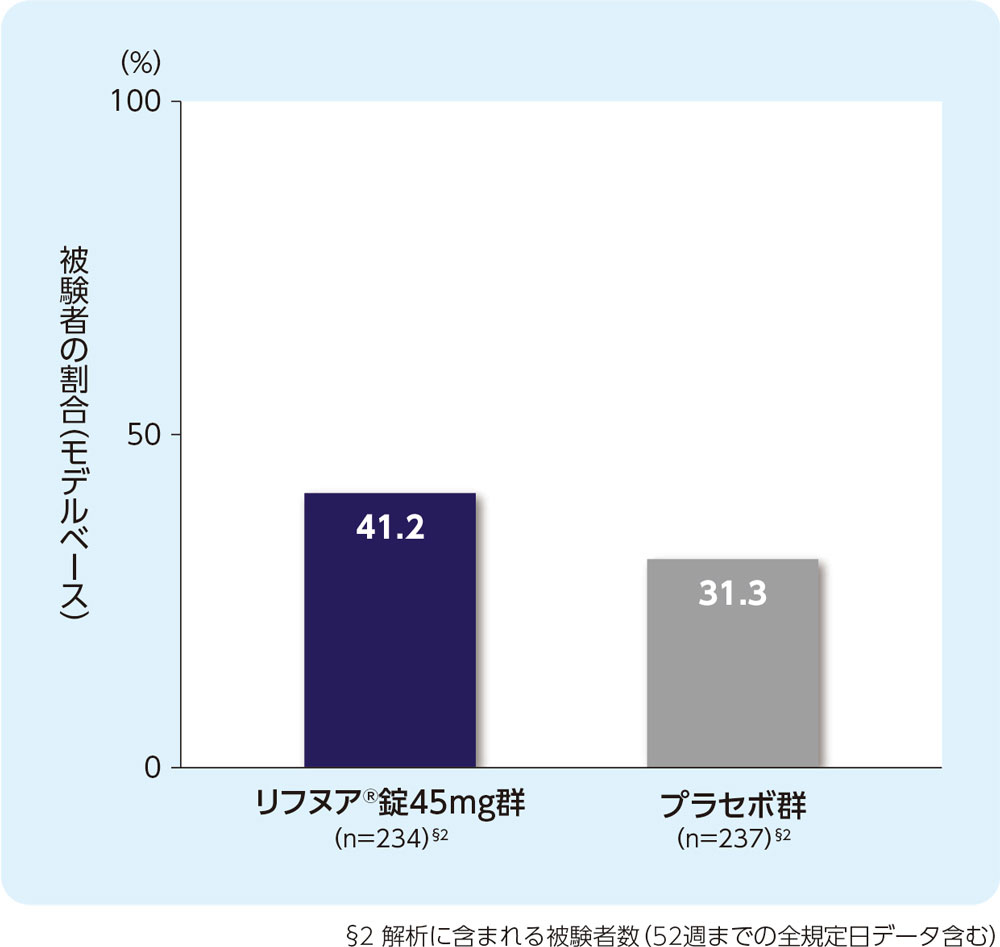
(5)12週時の咳重症度VASがベースラインから30mm以上減少した被験者の割合(FAS)[副次評価項目]
12週時の咳重症度VASがベースラインから30mm以上減少した被験者の割合は、リフヌア®錠45mg群で41.2%、プラセボ群で31.3%でした。
ベースライン値(平均値±標準偏差)
リフヌア®錠45mg群(n=243):67.9±12.83
プラセボ群(n=241):69.1±13.85
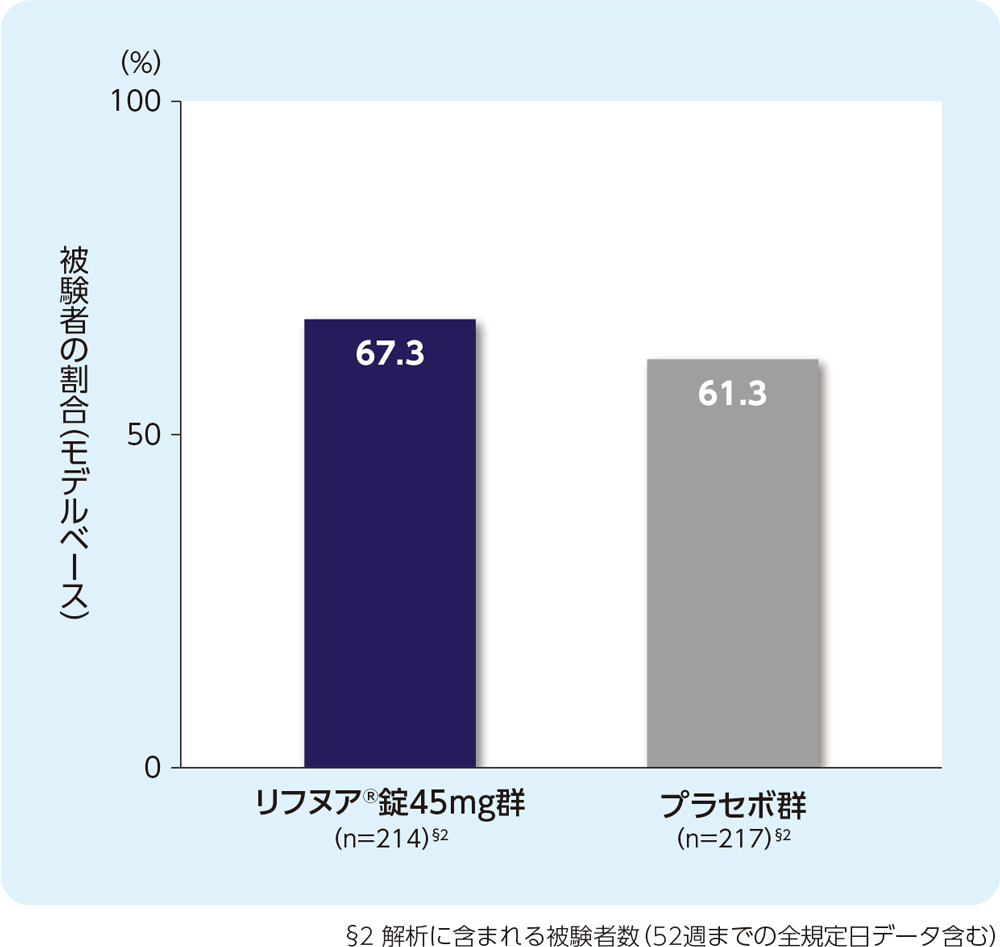
(6) 12週時のLCQ合計スコアがベースラインから1.3ポイント以上増加した被験者の割合(FAS)[副次評価項目]
12週時のLCQ合計スコアがベースラインから1.3ポイント以上増加した被験者の割合は、リフヌア®錠45mg群で67.3%、プラセボ群で61.3%でした。
ベースライン値(平均値±標準偏差)
リフヌア®錠45mg群(n=236):10.5±2.73
プラセボ群(n=229):10.0±3.08
(7)安全性 *[主要評価項目]
| リフヌア®錠45mg群 | リフヌア®錠15mg群 | プラセボ群 | ||
|---|---|---|---|---|
| 安全性評価例数 | 243 | 244 | 243 | |
| 有害事象* | 発現例数(%) | 208(85.6) | 186(76.2) | 184(75.7) |
| 副作用 | 発現例数(%) | 158(65.0) | 49(20.1) | 47(19.3) |
| 投与中止に至った 有害事象* |
発現例数(%) | 51(21.0) | 15( 6.1) | 14( 5.8) |
主な副作用(いずれかの投与群で発現率1%以上)
| 副作用の種類 | リフヌア®錠45mg群 | リフヌア®錠15mg群 | プラセボ群 | |||
|---|---|---|---|---|---|---|
| 発現例数 | 発現率(%) | 発現例数 | 発現率(%) | 発現例数 | 発現率(%) | |
| 胃腸障害 | ||||||
| 下痢 | 3 | 1.2 | 2 | 0.8 | 3 | 1.2 |
| 口内乾燥 | 13 | 5.3 | 3 | 1.2 | 6 | 2.5 |
| 消化不良 | 3 | 1.2 | 1 | 0.4 | 1 | 0.4 |
| 口の感覚鈍麻 | 10 | 4.1 | 2 | 0.8 | 0 | 0.0 |
| 悪心 | 9 | 3.7 | 1 | 0.4 | 5 | 2.1 |
| 口の錯感覚 | 8 | 3.3 | 3 | 1.2 | 1 | 0.4 |
| 流涎過多 | 4 | 1.6 | 0 | 0.0 | 0 | 0.0 |
| 一般・全身障害及び投与部位の状態 | ||||||
| 倦怠感 | 3 | 1.2 | 0 | 0.0 | 0 | 0.0 |
| 口渇 | 3 | 1.2 | 0 | 0.0 | 0 | 0.0 |
| 感染症及び寄生虫症 | ||||||
| 上咽頭炎 | 0 | 0.0 | 0 | 0.0 | 3 | 1.2 |
| 臨床検査 | ||||||
| 尿中結晶陽性 | 4 | 1.6 | 3 | 1.2 | 5 | 2.1 |
| 体重増加 | 4 | 1.6 | 0 | 0.0 | 0 | 0.0 |
| 代謝及び栄養障害 | ||||||
| 食欲減退 | 8 | 3.3 | 1 | 0.4 | 1 | 0.4 |
| 神経系障害 | ||||||
| 味覚消失 | 33 | 13.6 | 2 | 0.8 | 0 | 0.0 |
| 浮動性めまい | 2 | 0.8 | 3 | 1.2 | 0 | 0.0 |
| 味覚不全 | 88 | 36.2 | 20 | 8.2 | 7 | 2.9 |
| 頭痛 | 9 | 3.7 | 5 | 2.0 | 2 | 0.8 |
| 味覚過敏 | 3 | 1.2 | 0 | 0.0 | 0 | 0.0 |
| 味覚減退 | 13 | 5.3 | 5 | 2.0 | 1 | 0.4 |
| 味覚障害 | 23 | 9.5 | 2 | 0.8 | 1 | 0.4 |
| 呼吸器、胸郭及び縦郭障害 | ||||||
| 咽喉乾燥 | 3 | 1.2 | 2 | 0.8 | 0 | 0.0 |
| 口腔咽頭痛 | 4 | 1.6 | 3 | 1.2 | 0 | 0.0 |
| 皮膚及び皮下組織障害 | ||||||
| 丘疹性皮疹 | 2 | 0.8 | 0 | 0.0 | 3 | 1.2 |
投与中止に至った副作用は、リフヌア®錠45mg群では43例(味覚不全15例、味覚消失11例、味覚障害3例、咳嗽、口内乾燥、口の錯感覚が各2例、浮動性めまい、体重減少、舌苔、胸部不快感、呼吸困難、異常感、頭痛、咽喉刺激感、口の感覚鈍麻、舌不快感、舌そう痒症、薬物不耐性、高粘稠性気管支分泌物、腹痛、上腹部痛、発疹、トランスアミナーゼ上昇、胃炎、筋痙縮、悪心、嘔吐、食欲減退、倦怠感が各1例)、リフヌア®錠15mg群では6例(味覚不全2例、胃食道逆流性疾患、過敏症、嘔吐、下痢が各1例)、プラセボ群では7例(下痢、そう痒症、発疹、頭痛、尿管結石症、咳嗽、片頭痛、高血圧クリーゼが各1例)でした。
本試験において重篤な副作用は、リフヌア®錠45mg群及び15mg群では認められず、プラセボ群では尿管結石症の1例でした。
本試験における死亡例はリフヌア®錠15mg群で気道感染が1例、プラセボ群で死亡及び偶発的死亡が各1例認められ、いずれも治験薬との因果関係は否定されました。
※味覚に関連する有害事象の詳細は、030試験と併合した結果を海外第Ⅲ相試験ページに掲載
- 6.
-
用法及び用量
通常、成人にはゲーファピキサントとして1回45mgを1日2回経口投与する。
臨床試験で用いたPRO評価及びレスポンダーの定義
24時間の咳嗽頻度(1時間あたりの回数)がベースラインから30%以上減少した被験者の割合
レスポンダーの定義として用いた臨床的に重要な改善の閾値(30%以上減少)は、海外第Ⅱ相試験(012試験)の結果に基づく分布法及びアンカー法を組み合わせた解析4)により、24時間の咳嗽頻度がベースラインから30%以上減少した場合にPGIC(全般的な変化への影響を評価)の改善が予測されることから設定した。
CSD合計スコアの週平均がベースラインから1.3ポイント以上又は
2.7ポイント以上減少した被験者の割合
咳重症度日誌(CSD)は咳嗽頻度(3項目)、咳嗽の強度(2項目)及び咳嗽による支障(2項目)を0~10の11段階で評価する質問票で、スコアが高いほど重症であることを示す。
本試験におけるレスポンダーの定義に用いた1.3及び2.7ポイント以上の減少は、海外第Ⅱ相試験(012試験)のPGICの解析結果5)に基づき設定した。
咳重症度VASがベースラインから30mm以上減少した被験者の割合
咳重症度VASは、0(咳をしない)、100(極めてひどい咳)の範囲に対応した100mmのVASを用いて、過去24時間の咳重症度を評価する。
レスポンダーの定義に用いた30mm以上の減少は、海外第Ⅱ相試験(012試験)のPGICの解析結果6)に基づき設定した。
LCQ合計スコアがベースラインから1.3ポイント以上増加した被験者の割合
LCQは3つの個別の領域(身体的、精神的機能、社会的)に分類される19項目の質問で構成されているQOL質問票である。
各質問は、1~7ポイントの7段階のリッカート尺度で、評価前2週間の症状及び症状がQOLに及ぼす影響を評価する。3つの個別領域における質問を平均したスコアを合計した3~21ポイントをLCQ合計スコアとして評価する。スコアが高いほどQOLが良好であることを示す。
レスポンダーの定義に用いた1.3ポイント以上の増加はLCQに関する公表データ7)及び海外第Ⅱ相試験(012試験)データ8)に基づき、臨床的に重要なスコアの改善と考えられたことから設定した。
PGICについて
「非常に大幅に改善された」から「非常に大幅に悪化した」まで7段階のリッカート尺度を用いて、治験開始時と比較した状態の変化に対する影響を評価する質問票であり、異なる評価項目のレスポンダーの定義として臨床的に重要な改善の閾値を推定する解析にも使用される。海外第Ⅱ相試験(012試験)のPGICの解析には、PGICの改善を示す3つのカテゴリー(非常に大幅に改善された、非常に改善された、最小限に改善された)を用いた。
- 1)承認時評価資料:国際共同第Ⅲ相試験(027試験)
- 2)McGarvey LP, et al. Lancet. 2022; 399(10328): 909-23.
【利益相反】本試験はMSD社の支援により実施された。 - 3)Irwin RS, et al. Chest. 2006; 129(1 Suppl): 1S-23S.
-
4)Schelfout J, et al. Lung. 2022; 200(6): 717-24.
【利益相反】本研究はMSD社の支援により実施され、著者のうち4名が同社の社員である。 -
5)Nguyen AM, et al. Ther Adv Respir Dis. 2020; 14: 1-15.
【利益相反】本研究はMSD社の支援により実施され、著者のうち1名が同社の社員である。 -
6)Nguyen AM, et al. Ther Adv Respir Dis. 2021; 15: 1-13.
【利益相反】本研究はMSD社の支援により実施され、著者のうち4名が同社の社員である。 - 7)Raj AA, et al. Handb Exp Pharmacol. 2009;(187): 311-20.15)
-
8)Nguyen AM, et al. Ther Adv Respir Dis. 2022; 16: 1-13.
【利益相反】本研究はMSD社の支援により実施され、著者のうち4名が同社の社員である。
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。