

-臨床成績-
国内第Ⅲ相多施設共同無作為化二重盲検プラセボ対照並行群間比較試験1)
本試験開始時に発行されていた間質性膀胱炎(IC)診療ガイドラインでは、ICはハンナ病変を有するハンナ型と、ハンナ病変は有さないが点状出血又は拡張後粘膜出血がある非ハンナ型とに分類されていたため、本試験はハンナ型及び非ハンナ型のいずれも組み入れ対象となっています。承認時評価資料であるため、承認外の非ハンナ型を含めた試験成績を記載しています。
試験概要
| 目的 |
主要目的: 日本人間質性膀胱炎患者を対象として、投与後12週のO'Leary & Sant間質性膀胱炎症状スコア(ICSI)のベースラインからの変化量について、ジムソ群のプラセボ群に対する優越性を検証する。 副次的目的: 副次的評価項目について、ジムソ群のプラセボ群に対する優越性を検討する。また、日本人間質性膀胱炎患者を対象として、ジムソ®内注入液50%を2週間間隔で6回膀胱内注入した際の安全性を検討する。 |
|---|---|
| 対象 |
日本人間質性膀胱炎患者* 96例
|
| 試験デザイン |
多施設共同無作為化二重盲検プラセボ対照並行群間比較試験 |
| 方法 |
観察期(2週間)にプラセボ50mLを単回膀胱内注入した。ICSIスコアを割付け因子とした動的割付けにより、被験者をジムソ群又はプラセボ群のいずれかに1:1の比で無作為に割付け、それぞれジムソ®膀胱内注入液50%50mL又はプラセボ50mLを2週間間隔で6回膀胱内注入した。投与方法は、経尿道的に膀胱内に挿入されたカテーテルから、治験薬を膀胱内に注入し、可能な限り15分間以上膀胱内に保持させた後、自然排尿させることとした。 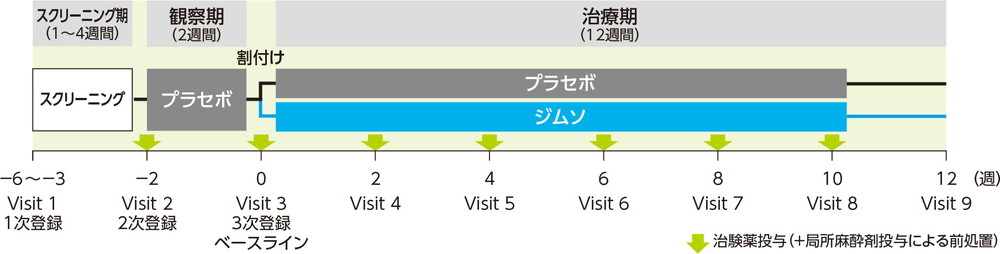
|
| 評価項目 |
有効性評価項目:
|
| 解析計画 |
有効性の主たる解析対象集団はFASとした。 主要評価項目: MMRM(Mixed Model for Repeated Measures)を用いてジムソ群のプラセボ群に対する最小二乗平均の差及びその両側95%信頼区間を推定し、最小二乗平均の差の検定(有意水準両側5%)を行うことで、ジムソ群のプラセボ群に対する優越性を検証した。なお、MMRMでは固定効果として測定時期、投与群及び投与群と測定時期の交互作用項、共変量としてベースラインのICSI値を設定した。 副次的評価項目: 投与後12週における各項目のベースラインからの変化量は、MMRMを用いて主要評価項目と同様の方法でジムソ群のプラセボ群に対する優越性を検討した。投与後12週におけるGRAは、投与群別に、GRA(1~7)、GRAレスポンダー(GRA1又はGRA2)及びノンレスポンダー(GRA3~GRA7)について、それぞれ被験者数及び割合(%)を集計、GRAレスポンダーの割合の群間差及びその95%信頼区間を算出し、フィッシャーの正確確率検定を用いて割合の群間差の検定を行った。 その他の有効性評価項目: 主要評価項目及び副次的評価項目でMMRMを用いて実施した統計解析の4週及び8週の結果を、12週と同様に表示した。 |
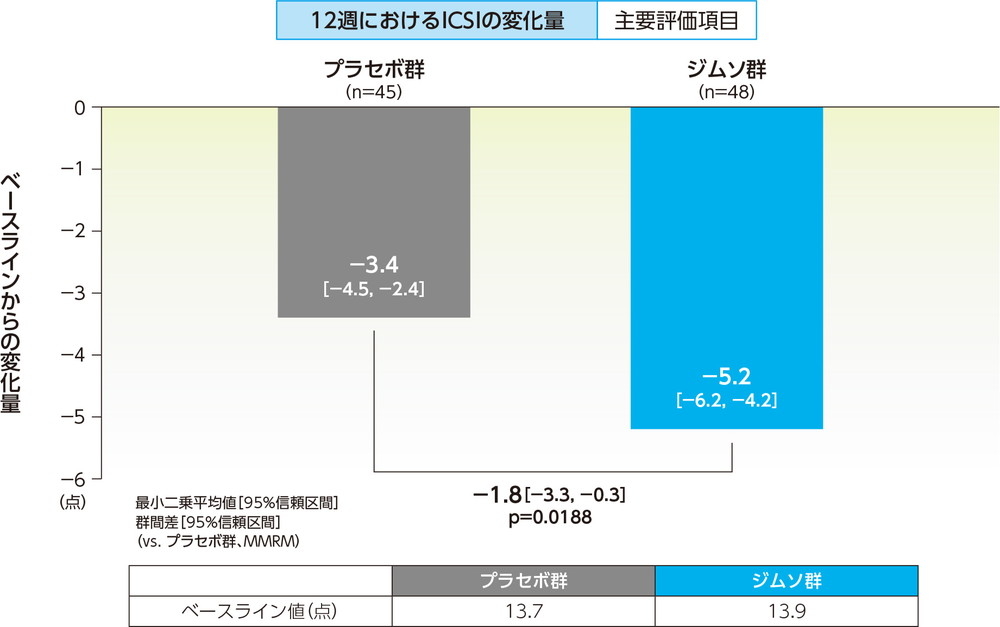
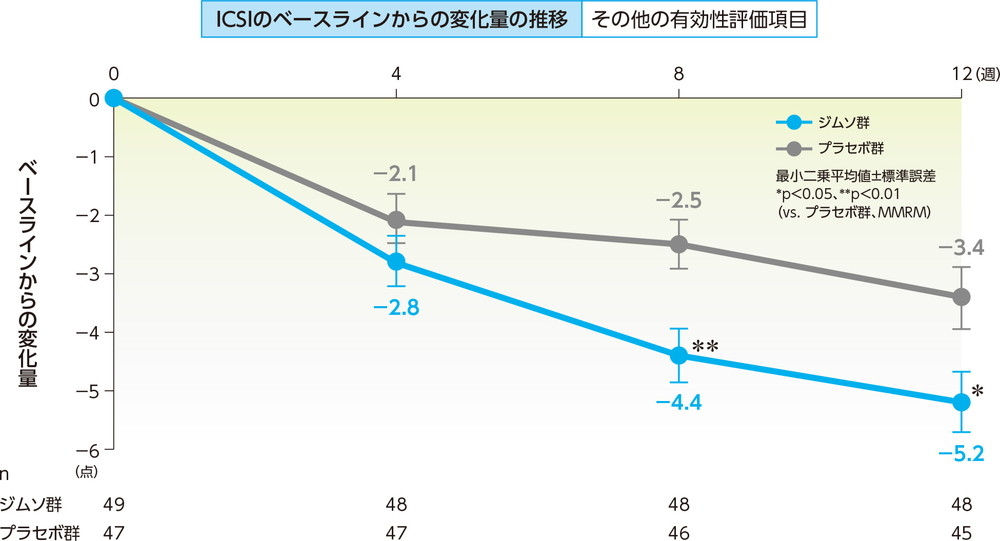
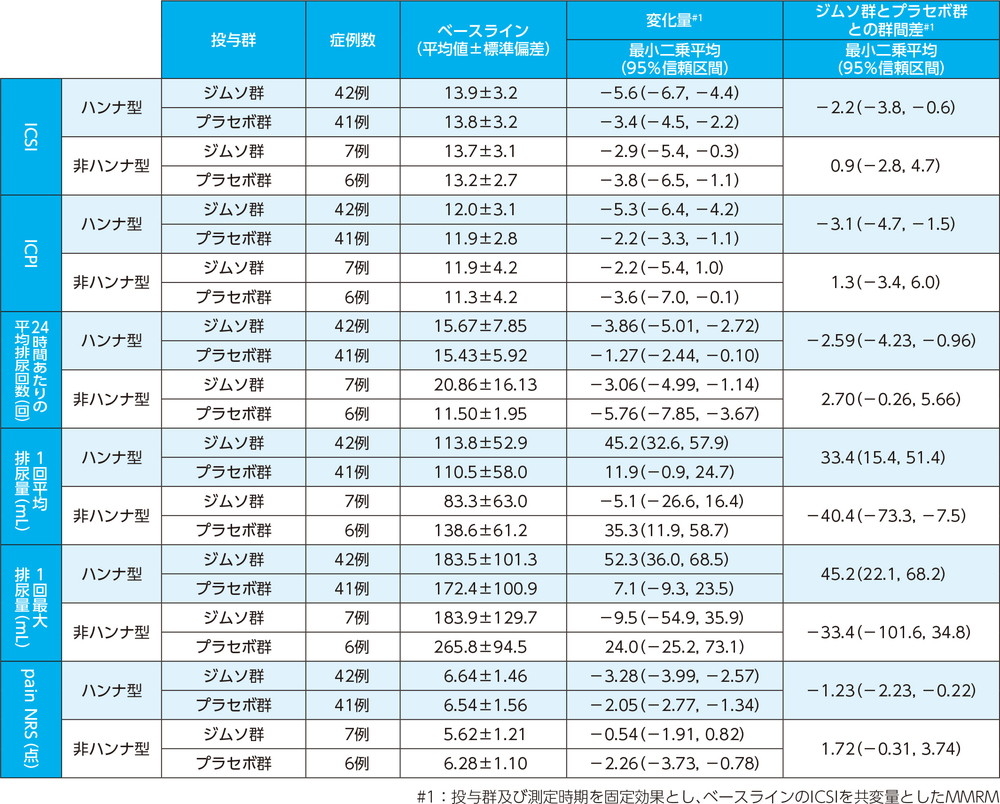
ICSIのベースラインからの変化量(12週、FAS)主要評価項目 その他の有効性評価項目
12週でのICSIのベースラインからの変化量の最小二乗平均は、ジムソ群で−5.2点、プラセボ群で−3.4点であった。
ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は−1.8点(−3.3点, −0.3点)であり、ジムソ群のプラセボ群に対する優越性が検証された(p=0.0188、MMRM、共変量=ベースライン値(0週))。
ICPIのベースラインからの変化量(12週、FAS)副次的評価項目
12週でのICPIのベースラインからの変化量の最小二乗平均は、ジムソ群で−4.9点、プラセボ群で−2.4点であった。
ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は−2.5点(−4.0点, −1.0点)であり、ジムソ群はプラセボ群に対し有意な減少を示した(p=0.0014、MMRM、共変量=ベースライン値(0週))。

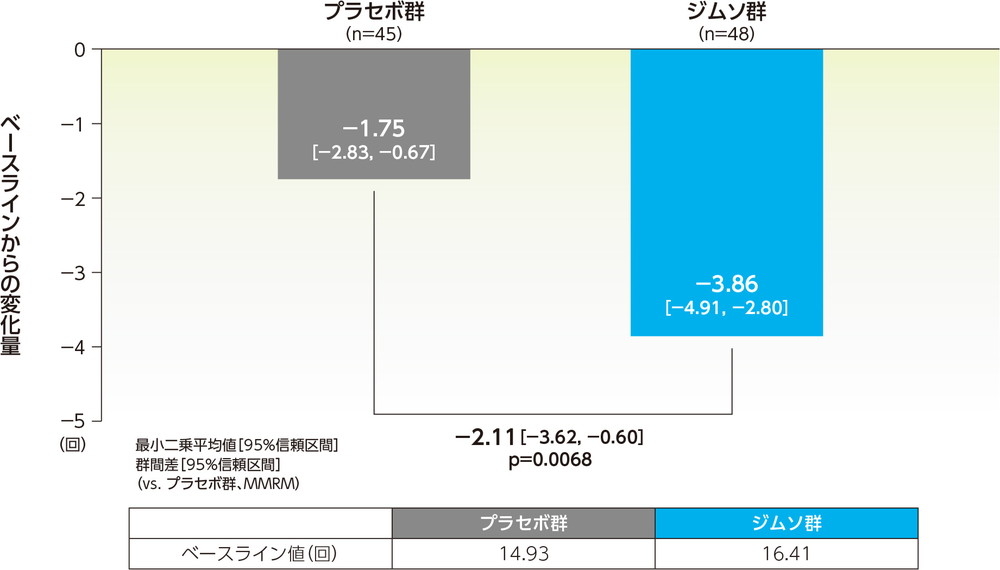
24時間あたりの平均排尿回数のベースラインからの変化量(12週、FAS)副次的評価項目
12週での24時間あたりの平均排尿回数のベースラインからの変化量の最小二乗平均は、ジムソ群で−3.86回、プラセボ群で−1.75回であった。ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は−2.11回(−3.62回, −0.60回)であり、ジムソ群はプラセボ群に対し有意な減少を示した(p=0.0068、MMRM、共変量=ベースライン値(0週))。
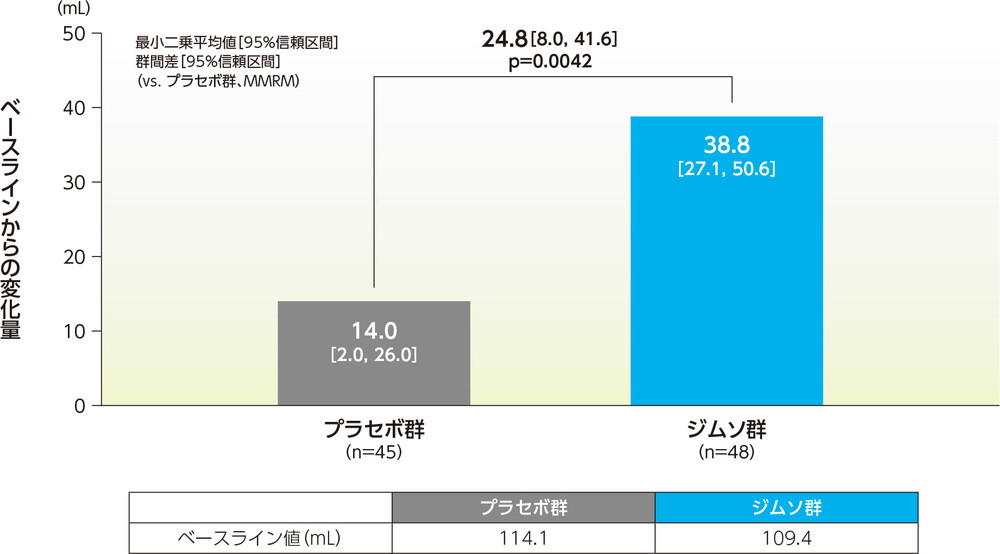
1回平均排尿量のベースラインからの変化量(12週、FAS)副次的評価項目
12週での1回平均排尿量のベースラインからの変化量の最小二乗平均は、ジムソ群で38.8mL、プラセボ群で14.0mLであった。ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は24.8mL(8.0mL, 41.6mL)であり、ジムソ群はプラセボ群に対し有意な増加を示した(p=0.0042、MMRM、共変量=ベースライン値(0週))。
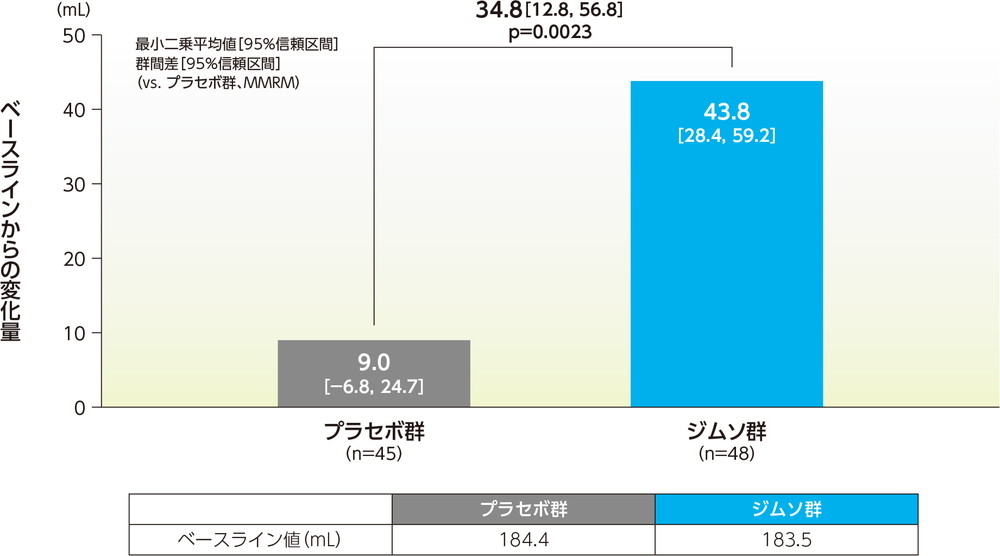
1回最大排尿量のベースラインからの変化量(12週、FAS)副次的評価項目
12週での1回最大排尿量のベースラインからの変化量の最小二乗平均は、ジムソ群で43.8mL、プラセボ群で9.0mLであった。ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は34.8mL(12.8mL, 56.8mL)であり、ジムソ群はプラセボ群に対し有意な増加を示した(p=0.0023、MMRM、共変量=ベースライン値(0週))。
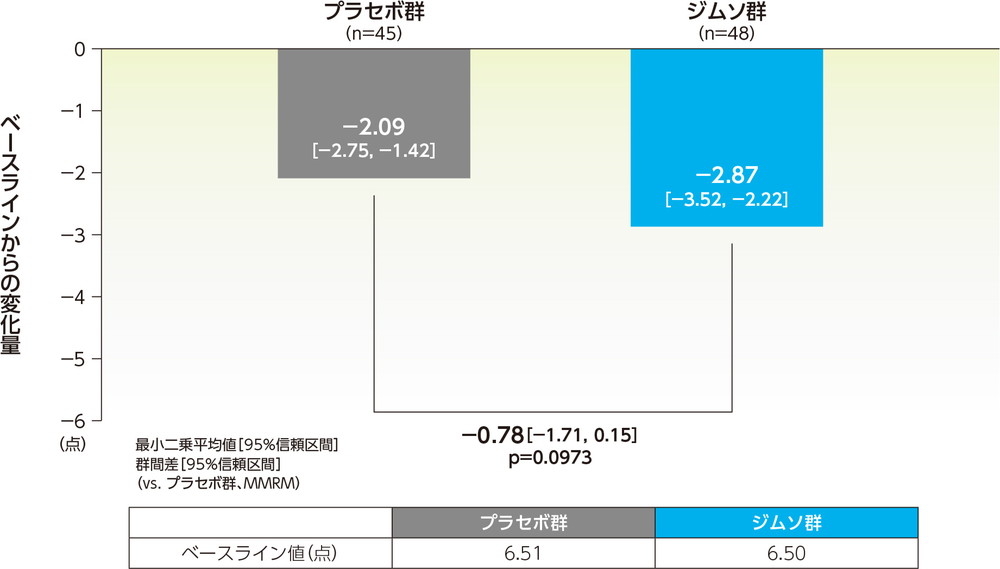
pain NRSのベースラインからの変化量(12週、FAS)副次的評価項目
12週でのpain NRSのベースラインからの変化量の最小二乗平均は、ジムソ群で−2.87点、プラセボ群で−2.09点であった。ジムソ群のプラセボ群に対する変化量の最小二乗平均の差(95%信頼区間)は−0.78点(−1.71点, 0.15点)であった(p=0.0973、MMRM、共変量=ベースライン値(0週))。
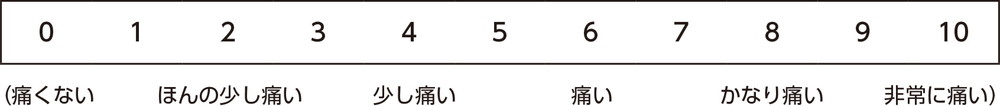
pain NRS(膀胱痛のNumerical rating scale:膀胱痛の数値化スケール)について
膀胱の痛みについて、「全くない」を0、想像できる最大の強さを10としたとき、昨日起床してから今日起きる前までに感じた一番強い痛みを患者自身が評価した。
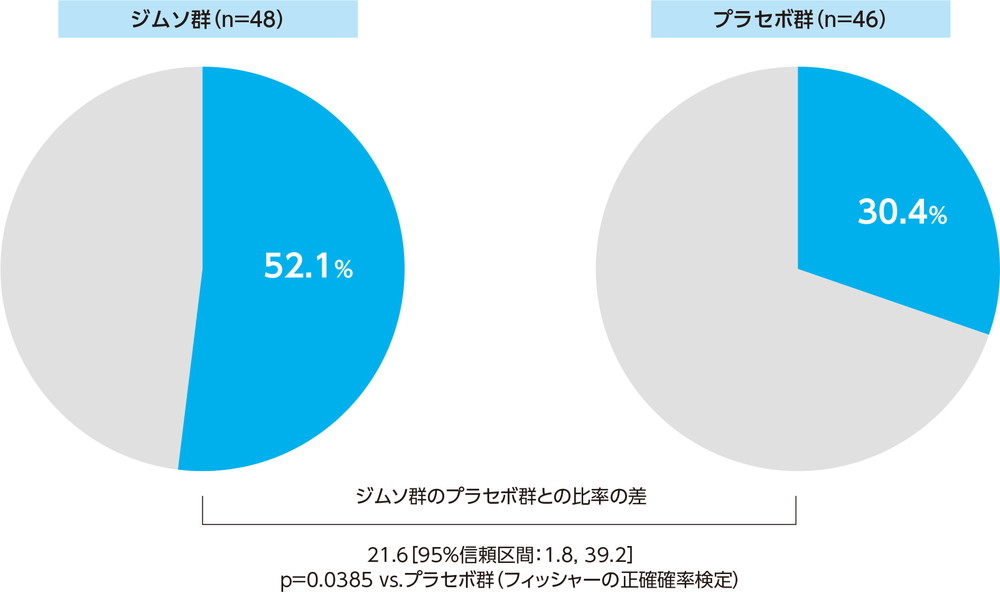
GRA(12週、FAS)副次的評価項目
12週でのGRAレスポンダーの割合は、ジムソ群で52.1%(25/48例)、プラセボ群で30.4%(14/46例)であった。
ジムソ群のプラセボ群に対するGRAレスポンダーの割合の群間差(95%信頼区間)は21.6%(1.8%, 39.2%)であり、ジムソ群はプラセボ群に対し有意に高かった(p=0.0385、フィッシャーの正確確率検定)。
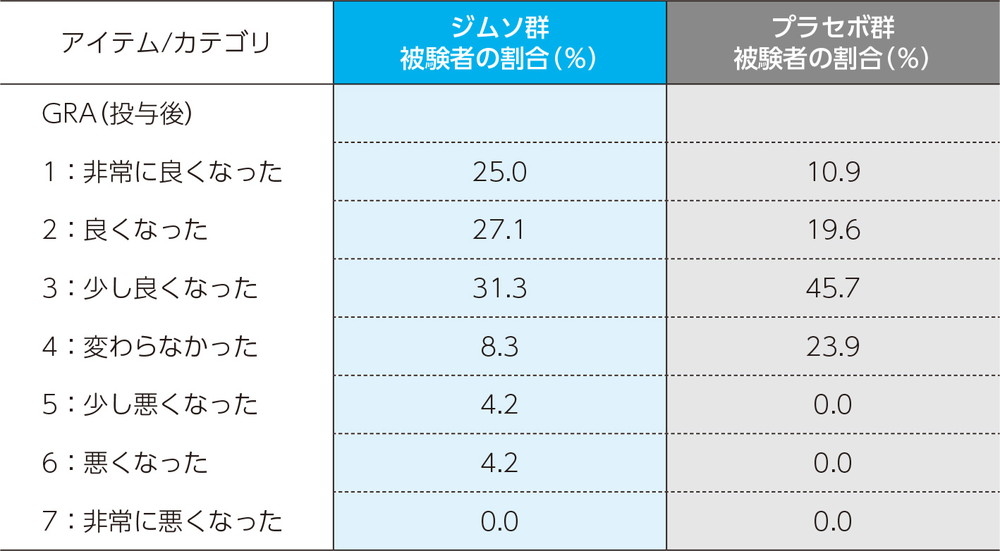
GRA(Global response assessment:全般的改善度評価)について
全般的な『印象』に関する質問。本試験の開始前に比べて膀胱の状態がどのように変化したのか、7段階で患者自身が評価した。
GRAにおいて「1 : 非常に良くなった」及び「2 : 良くなった」と評価した被験者をGRAレスポンダー、それ以外の被験者(欠測を除く)をノンレスポンダーと定義する。
- 4.
-
効能又は効果
間質性膀胱炎(ハンナ型)の諸症状(膀胱に関連する慢性の骨盤部の疼痛、圧迫感及び不快感、尿意亢進又は頻尿等の下部尿路症状)の改善
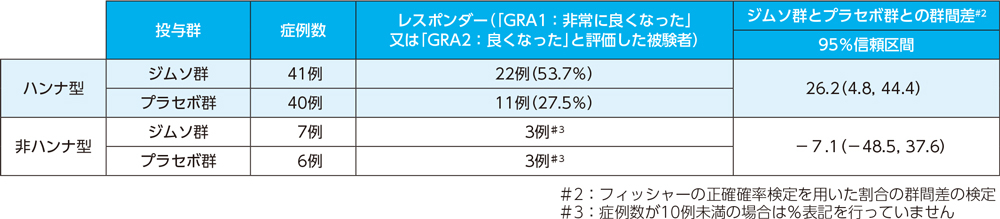
部分集団解析(ハンナ型及び非ハンナ型、12週、FAS)副次的評価項目
部分集団におけるMMRM解析は、当初計画されたものではなく承認審査の過程において実施されました。承認時評価資料であり、電子添文17. 臨床成績の項へ全体集団に加えてハンナ型集団及び非ハンナ型集団の結果の記載が求められたため本資材にも掲載しています。
投与12週における全般的改善度評価(GRA)レスポンダーの割合
- 4.
-
効能又は効果
間質性膀胱炎(ハンナ型)の諸症状(膀胱に関連する慢性の骨盤部の疼痛、圧迫感及び不快感、尿意亢進又は頻尿等の下部尿路症状)の改善
- 5.
-
効能又は効果に関連する注意
本剤を投与する際、十分な問診により臨床症状を確認するとともに、類似の症状を呈する疾患(尿路性器感染症、尿路結石、膀胱癌や前立腺癌などの下部尿路における新生物、過活動膀胱や前立腺肥大症等)があることに留意し、膀胱内視鏡、尿検査等により除外診断を実施すること。その上で、膀胱内視鏡検査によりハンナ病変が認められ、間質性膀胱炎(ハンナ型)の確定診断を受けた患者にのみ投与すること。
安全性
副作用発現率
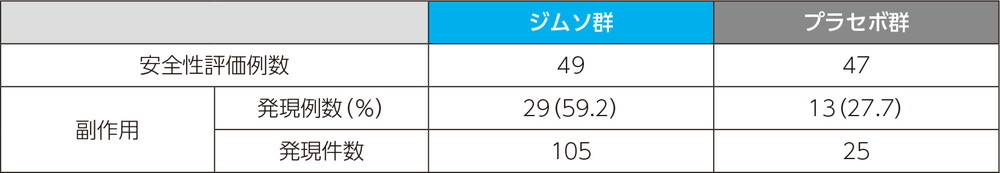
主な副作用(いずれかの投与群で発現例数3例以上)
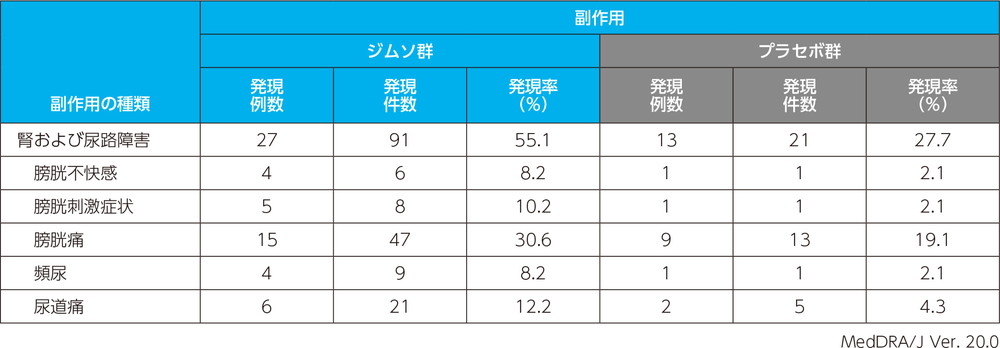
投与中止に至った副作用は、プラセボ群で発疹が1例1件であった。本試験においては死亡例、重篤な副作用は認められなかった。
注目すべき有害事象
投与時反応に関する有害事象
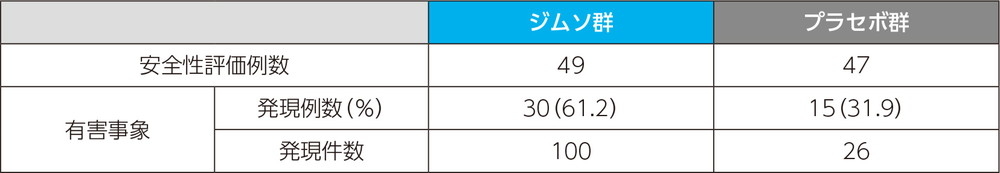
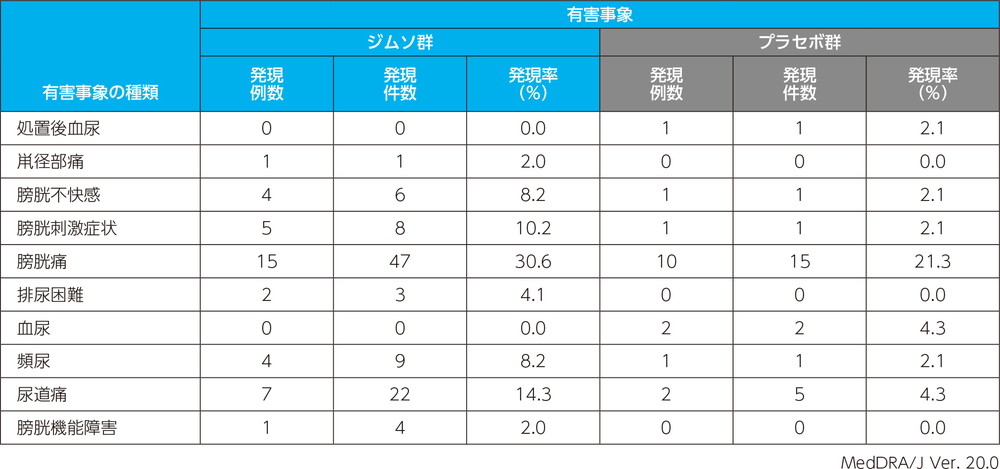
・投与時反応に関する有害事象の治験薬投与回数別の発現割合
ジムソ群及びプラセボ群ともに初回投与後の発現割合が最も高く、それぞれ53.1%(26/49例)及び23.4%(11/47例)であった。
・投与時反応に関する有害事象の各治験薬投与日から発現日までの経過日数
中央値は、ジムソ群及びプラセボ群ともにいずれの投与回数でも1日であった。また、投与時反応に関する有害事象の発現から転帰確認までの経過日数の全投与回数での中央値は、ジムソ群では2日、プラセボ群では1日であった。
- 8.
-
重要な基本的注意
投与時反応(膀胱痛、尿道痛、膀胱刺激症状、膀胱不快感等)があらわれることがあるため、必要に応じて、本剤投与前に局所麻酔剤の膀胱内注入を行った上で投与すること。
呼気・皮膚の異常臭に関する有害事象
呼気・皮膚の異常臭に関する有害事象の発現割合はジムソ群で6.1%(3/49例)であり、呼気臭が2例3件、皮膚臭異常が1例1件であった。呼気・皮膚の異常臭に関する有害事象はいずれも治験責任医師等により治験薬との因果関係が否定できないと判断された。
水晶体障害に関する有害事象
呼気・皮膚本試験においては水晶体障害に関する有害事象は認められなかった。
- 15.
- その他の注意
- 15.2
- 非臨床試験に基づく情報(抜粋)
- 15.2.1
- ラット(9.9g/kg/日、経口)及びイヌ(1.1、3.3、9.9g/kg/日、経口)の反復投与毒性試験において、ラットでは投与18箇月後、イヌでは1.1g/kg群で投与31週後に、3.3及び9.9g/kg群では投与10週後に眼の変化(水晶体の混濁又は屈折率の変化)が認められたとの報告がある。
- 1) 承認時評価資料:日本人間質性膀胱炎患者を対象とした多施設共同、無作為化、二重盲検、プラセボ対照、並行群間比較試験(KRP116D-L301試験)
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。