

-臨床成績 皮膚疾患に伴うそう痒-
国内第Ⅲ相非盲検長期投与試験(湿疹・皮膚炎及び皮膚そう痒症)1)
| 目的 |
湿疹・皮膚炎及び皮膚そう痒症患者を対象としたデザレックス®の2週後の有効性並びに8~12週間投与時の安全性及び忍容性の検討 |
|---|---|
| 対象 |
12歳以上の湿疹・皮膚炎患者 65例、皮膚そう痒症患者 29例
<選択基準> |
| 試験デザイン |
多施設共同、非盲検、第Ⅲ相長期投与試験 |
| 試験方法 |
2週間以内の観察期間後、デザレックス®5mgを非盲検下で1日1回夕方に経口投与した。なお、投与開始後4週時以降8週時までの期間に10mg(5mg錠×2錠1日1回夕方投与)への増量を可とし、増量後4~8週間経口投与した(減量可)。 |
| 評価項目 |
<有効性>
副次評価項目:
<安全性> |
| 解析計画 |
主要評価項目及び副次評価項目は、経時測定データ解析モデル(時点、疾患群、時点と疾患群の交互作用を因子とし、時点はカテゴリ変数とした)を用いて評価した。 ※本剤の承認された用法・用量は1日1回5mgである。 |
痒みのスコアの判定基準

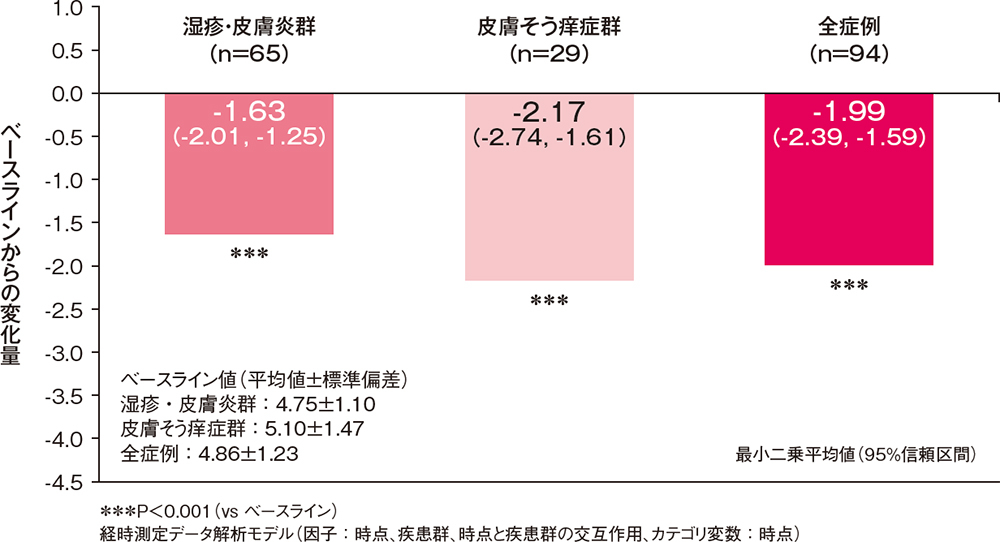
投与2週後の痒みスコアの合計のベースラインからの変化量[主要評価項目]
投与2週後の痒みスコア(日中の症状及び夜間の症状)の合計のベースラインからの変化量は、湿疹・皮膚炎群、皮膚そう痒症群ともにベースラインに比べて有意な低下を示しました(P<0.001、制約付き経時測定データ解析モデル)。
痒みスコア(日中の症状及び夜間の症状)の合計のベースラインからの変化量(FAS)
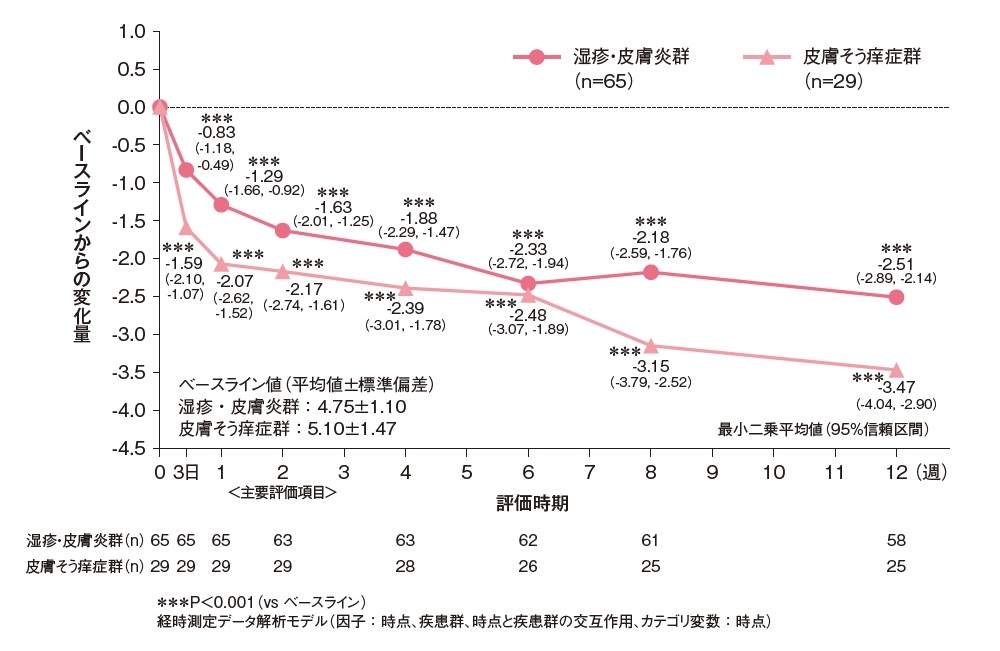
痒みスコアの合計のベースラインからの変化量[副次評価項目]
各評価時点の痒みスコア(日中の症状及び夜間の症状)の合計のベースラインからの変化量は、湿疹・皮膚炎群、皮膚そう痒症群のいずれも投与3日時点で有意に低下し、12週まで維持されました(P<0.001、制約付き経時測定データ解析モデル)。
痒みスコア(日中の症状及び夜間の症状)の合計のベースラインからの変化量(FAS)
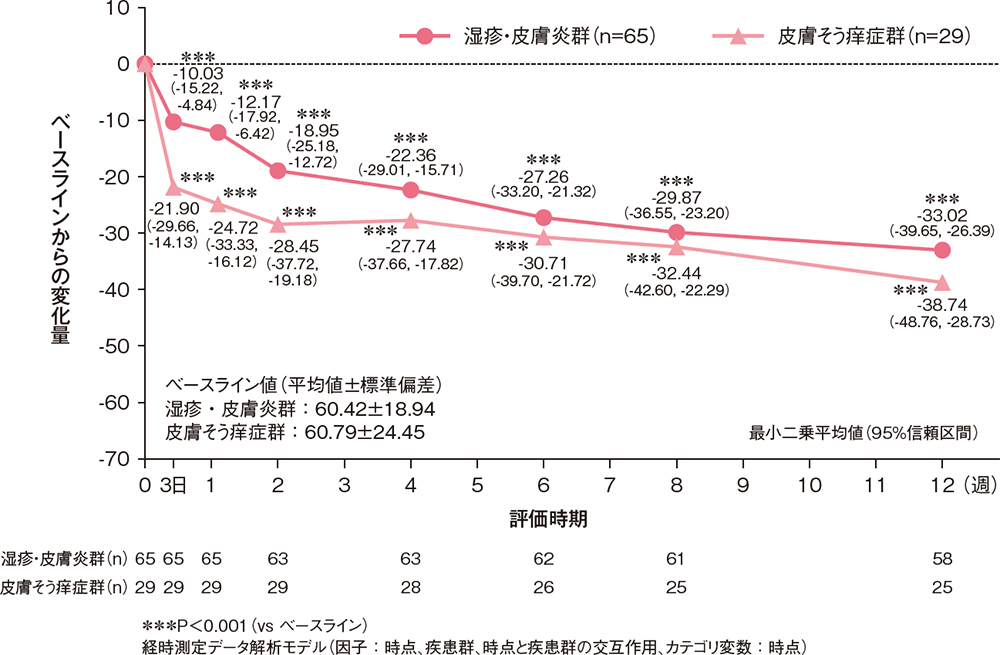
痒みの程度(VAS)のベースラインからの変化量[副次評価項目]
各評価時点において、VAS(0~100mm)で評価した痒みの程度のベースラインからの変化量は、湿疹・皮膚炎群、皮膚そう痒症群のいずれも投与3日時点で有意に低下し、12週までの各時点でも有意に低下していました(P<0.001、経時測定データ解析モデル)。
痒みの程度のベースラインからの変化量(FAS)
安全性
有害事象の発現率は、全体で52.1%(49/94例)、湿疹・皮膚炎群で53.8%(35/65例)、皮膚そう痒症群で48.3%(14/29例)でした。主な有害事象は、鼻咽頭炎[全体:18.1%(17/94例)、湿疹・皮膚炎群:20.0%(13/65例)、皮膚そう痒症群:13.8%(4/29例)]と傾眠[全体:4.3%(4/94例)、湿疹・皮膚炎群:6.2%(4/65例)、皮膚そう痒症群:0.0%(0/29例)]でした。このうち傾眠はすべて副作用と判断されましたが、いずれも治験薬投与中に回復が確認されました。
死亡は認められず、重篤な有害事象は湿疹・皮膚炎群の1例に入院を必要とする軽度の皮膚の新生物が認められました。その他の投与中止に至った有害事象は、湿疹・皮膚炎群のダニ皮膚炎、胃腸炎及び頭痛(いずれも各1例)、皮膚そう痒症群の皮脂欠乏性湿疹及び背部痛(いずれも各1例)でした。このうち、副作用と判定された事象は、皮脂欠乏性湿疹でした。
本試験では94例中66例(湿疹・皮膚炎群47例及び皮膚そう痒症群19例)が、投与4週目以降に10mg1日1回投与への増量を行いました。増量例及び非増量例別の有害事象発現率は、それぞれ51.5%(34/66例)、53.6%(15/28例)であり、発現率は増量例と非増量例で類似していました。
本試験で最も多く報告された副作用である傾眠は、増量例でより多く報告されましたが[増量例:4.5%(3/66例)、非増量例:3.6%(1/28例)]、発現時期はいずれも増量前でした。また、増量後に発現した副作用として、3例で臨床検査値異常の有害事象が認められ、そのうち肝酵素上昇が発現した1例では10mgから5mgに減量し、継続投与を行いました。
6.用法及び用量
通常、12歳以上の小児及び成人にはデスロラタジンとして1回5mgを1日1回経口投与する。8.重要な基本的注意
〈効能共通〉- 8.1
- 効果が認められない場合には、漫然と長期にわたり投与しないように注意すること。
- 1)承認時評価資料(皮膚疾患患者対象第Ⅲ相長期投与試験)
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。