

-臨床成績 蕁麻疹-
国内第Ⅲ相二重盲検プラセボ対照試験(慢性蕁麻疹)1)
| 目的 |
慢性蕁麻疹患者を対象としたプラセボに対するデザレックス®の有効性及び安全性の検討 |
|---|---|
| 対象 |
12歳以上の慢性蕁麻疹患者 239例 <選択基準> |
| 試験デザイン |
多施設共同、無作為化、プラセボ対照、第Ⅲ相二重盲検試験 |
| 試験方法 |
2週間以内の観察期間後、デザレックス®10mg群、デザレックス®5mg群又はプラセボ群の各群に1:1:1の比で無作為に割り付け、二重盲検下で2週間1日1回夕方経口投与した※。 |
| 評価項目 |
<有効性> 副次評価項目: <安全性> |
| 解析計画 |
主要評価項目は、デザレックス®10mg群及び5mg群のプラセボ群に対する優越性を検証した。 主要評価項目及び副次評価項目(全般改善度は除く)において、制約付き経時測定データ解析モデル(時点、時点と投与群、時点と年齢層、時点と重症度の交互作用を因子とした。なお、時点はカテゴリ変数として扱った)を用いて評価した。全般改善度は、改善率を反応変数とし、投与群、年齢層及び重症度を因子とするロジスティック回帰モデルを用いて評価した。 ※以下、主要評価項目及び副次評価項目についてはデザレックス®5mg群、プラセボ群の結果のみ記載、承認用量外の10mgはグラフから削除する。本剤の承認された用法・用量は、1日1回5mgである。 |
痒みのスコアの判定基準

発斑のスコアの判定基準
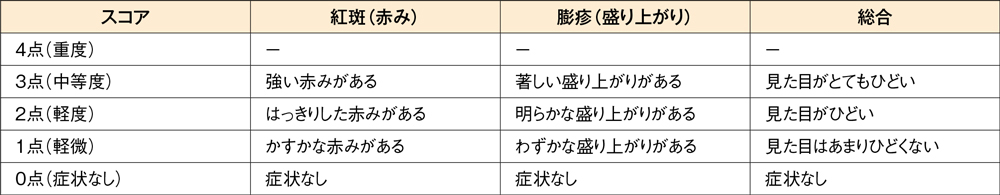
投与2週後の痒みスコアと発斑スコアの合計のベースラインからの変化量[主要評価項目]
投与2週後の痒みスコアと発斑スコアの合計のベースラインからの変化量はデザレックス®5mg群が-3.19、プラセボ群が-2.02、投与群間の差(95%信頼区間)は-1.17(-1.69,-0.65)であり、デザレックス®5mg群のプラセボ群に対する優越性が検証されました(P<0.001、制約付き経時測定データ解析モデル)。
痒みスコアと発斑スコアの合計のベースラインからの変化量(FAS)
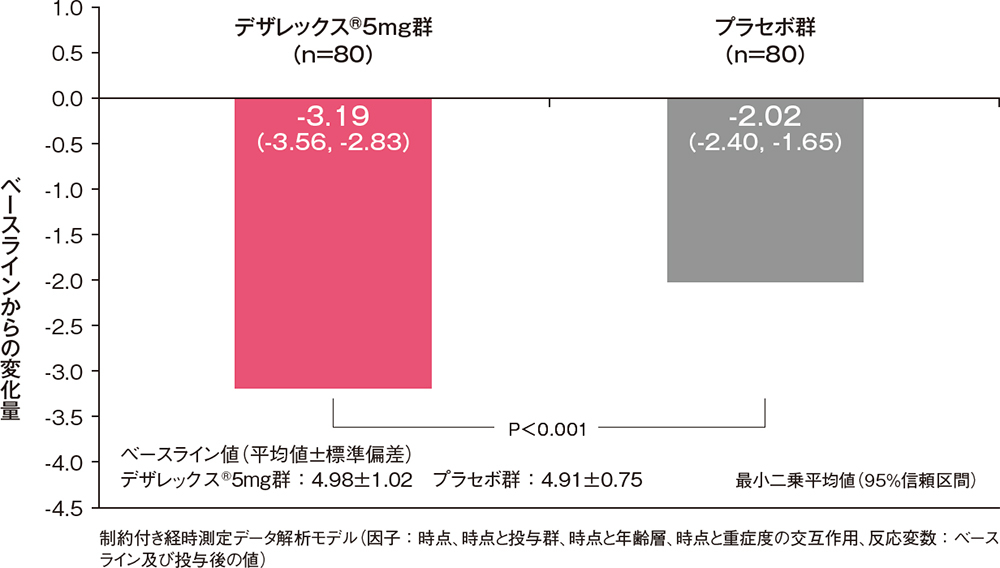
痒みスコアと発斑スコアの合計のベースラインからの変化量[副次評価項目]
痒みスコアと発斑スコアの合計のベースラインからの変化量は、投与3日後からデザレックス®5mg群がプラセボ群に比べ有意な低下を示しました(P<0.001、制約付き経時測定データ解析モデル)。
痒みスコアと発斑スコアの合計のベースラインからの変化量(FAS)
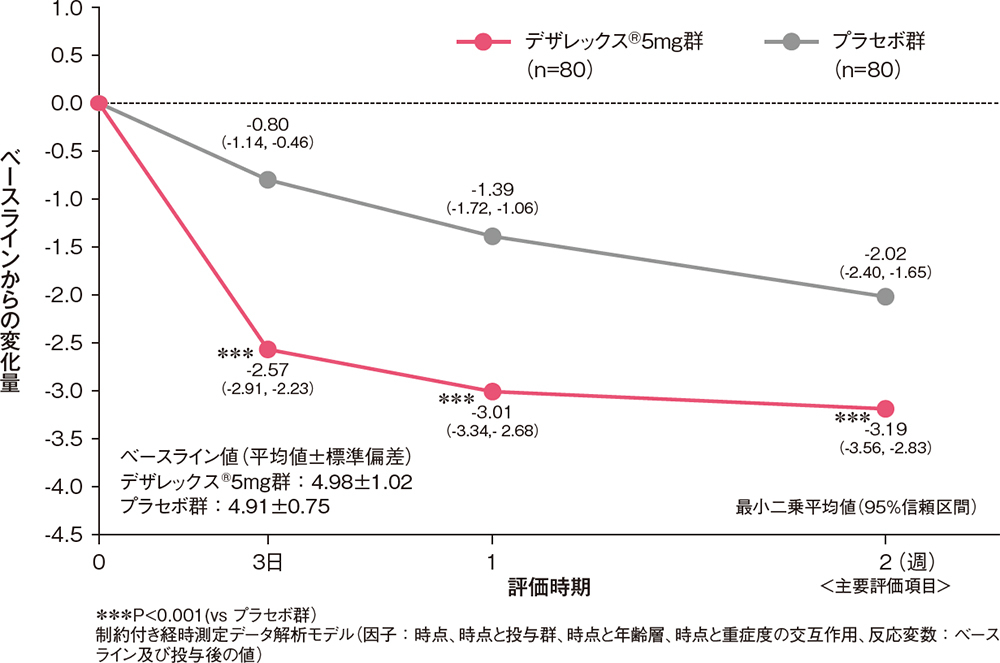
痒みスコアのベースラインからの変化量[副次評価項目]
痒みスコア(日中又は夜間の症状のうち程度の高い方)のベースラインからの変化量は、投与3日後からデザレックス®5mg群がプラセボ群と比較して有意な低下を示しました(P<0.001、制約付き経時測定データ解析モデル)。
痒みスコアのベースラインからの変化量(FAS)
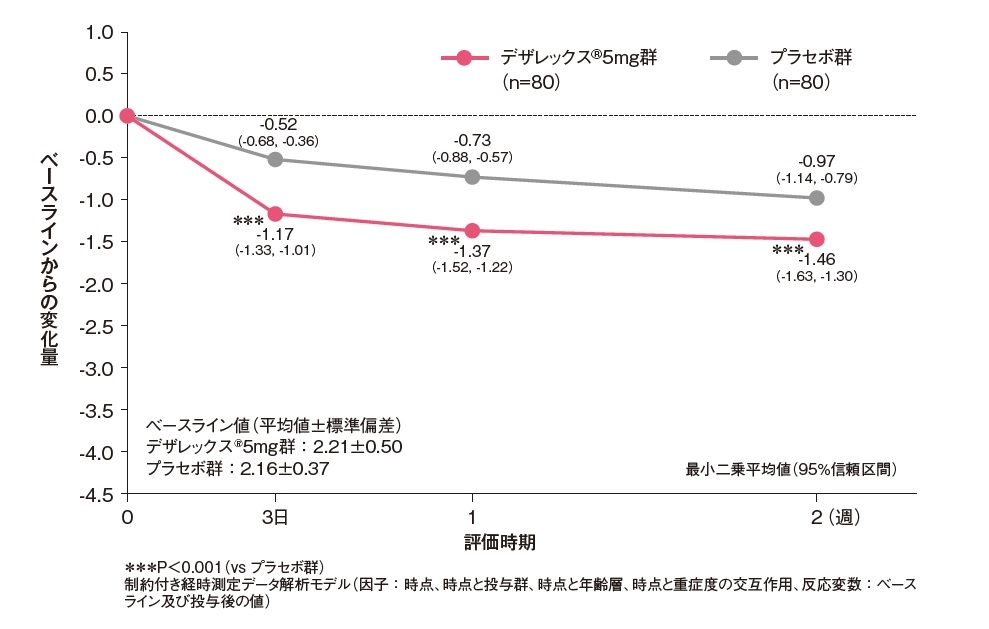
発斑スコアのベースラインからの変化量[副次評価項目]
発斑スコア(総合)のベースラインからの変化量は、投与3日後からデザレックス®5mg群がプラセボ群と比較して有意な低下を示しました(P<0.001、制約付き経時測定データ解析モデル)。
発斑スコアのベースラインからの変化量(FAS)
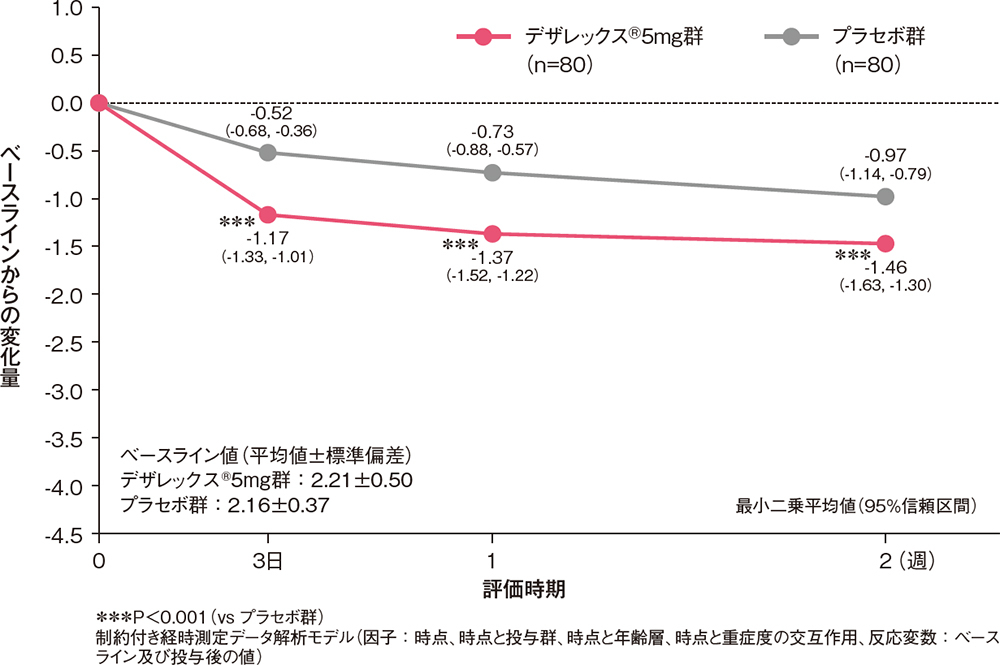
痒みの程度(VAS)のベースラインからの変化量[副次評価項目]
VAS(0~100mm)で評価した痒みの程度のベースラインからの変化量は、投与3日後からデザレックス®5mg群がプラセボ群と比較して有意な低下を示しました(P<0.001、制約付き経時測定データ解析モデル)。
痒みの程度(VAS)のベースラインからの変化量(FAS)
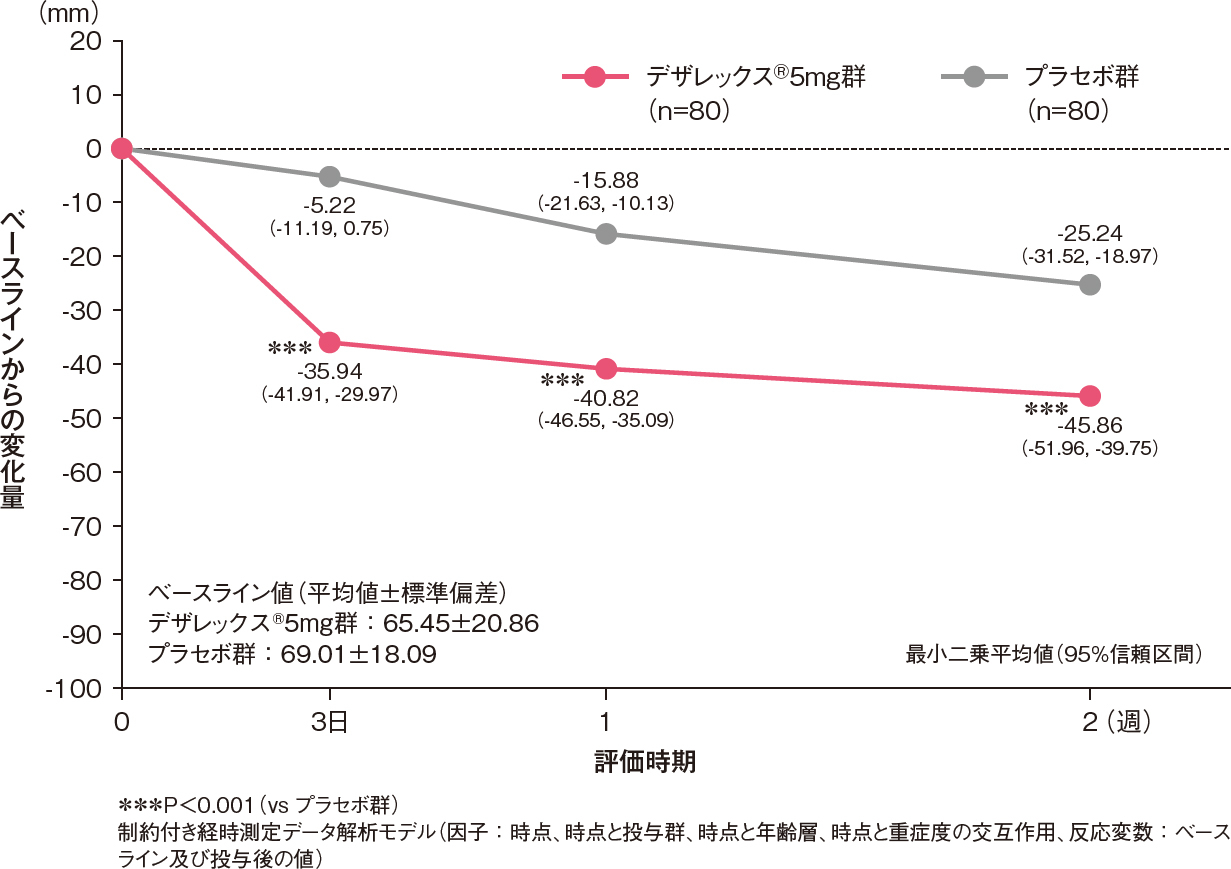
全般改善度の改善率(中等度改善以上の割合)[副次評価項目]
投与2週後における全般改善度の改善率(中等度改善以上の割合)は、デザレックス®5mg群がプラセボ群と比較して有意に高いものでした[プラセボ群に対するオッズ比:3.14、95%信頼区間:1.61-6.12、P=0.001、ロジスティック回帰モデル]。
投与2週後における全般改善度の改善率(中等度改善以上の割合)(FAS)
安全性
有害事象の発現率は、デザレックス®5mg群で30.0%(24/80例)、10mg群で22.8%(18/79例)及びプラセボ群で20.3%(16/79例)でした。デザレックス®5mg群又は10mg群で2%以上に発現した有害事象は、鼻咽頭炎[デザレックス®5mg群:10.0%(8/80例)、10mg群:3.8%(3/79例)、プラセボ群:2.5%(2/79例)]、傾眠[デザレックス®5mg群:3.8%(3/80例)、10mg群:6.3%(5/79例)、プラセボ群:3.8%(3/79例)]、口渇[デザレックス®5mg群:1.3%(1/80例)、10mg群:2.5%(2/79例)、プラセボ群:1.3%(1/79例)]及び頭痛[デザレックス®5mg群:0.0%(0/80例)、10mg群:2.5%(2/79例)、プラセボ群:1.3%(1/79例)]でした。
本試験で、死亡及びその他の重篤な有害事象はいずれの群でも認められませんでした。有害事象による投与中止はデザレックス®5mg群及び10mg群では認められず、プラセボ群で2.5%(2/79例)認められました。
副作用の発現率は、デザレックス®5mg群で8.8%(7/80例)、10mg群で13.9%(11/79例)及びプラセボ群で2.5%(2/79例)でした。副作用と判定された主な事象は傾眠[デザレックス®5mg群:3.8%(3/80例)、10mg群:6.3%(5/79例)、プラセボ群:2.5%(2/79例)]及び口渇[デザレックス®5mg群:1.3%(1/80例)、10mg群:2.5%(2/79例)、プラセボ群:0.0%(0/79例)]でした。
試験期間を通しての臨床検査値について、有害事象又は副作用と報告された事象は、γ-グルタミルトランスフェラーゼ増加(デザレックス®10mg群の1例)及び白血球数減少(デザレックス®5mg群の1例)でした。
6.用法及び用量
通常、12歳以上の小児及び成人にはデスロラタジンとして1回5mgを1日1回経口投与する。慢性特発性蕁麻疹患者に対する二重盲検比較試験(海外データ)2)
| 目的 |
慢性特発性蕁麻疹患者に対するデザレックス®の有効性及び安全性並びに効果発現時期の検討 |
|---|---|
| 対象 |
12歳以上の中等症から重症の慢性特発性蕁麻疹患者 190例 <選択基準> |
| 試験デザイン |
多施設共同無作為化二重盲検プラセボ対照比較試験 |
| 試験方法 |
デザレックス®5mg又はプラセボを1日1回、朝に6週間経口投与した。 |
| 評価項目 |
<有効性> 副次評価項目:痒みスコアの変化率(初回投与24時間後、1週間後及び6週間後の午前の平均値) 等 <安全性> |
| 解析計画 |
主要評価項目及び副次評価項目において、デザレックス®5mg群とプラセボ群の痒みスコアの変化率をtwo-way ANOVAを用いて評価し、デザレックス®5mg群のプラセボ群に対する優越性を検証した。 |
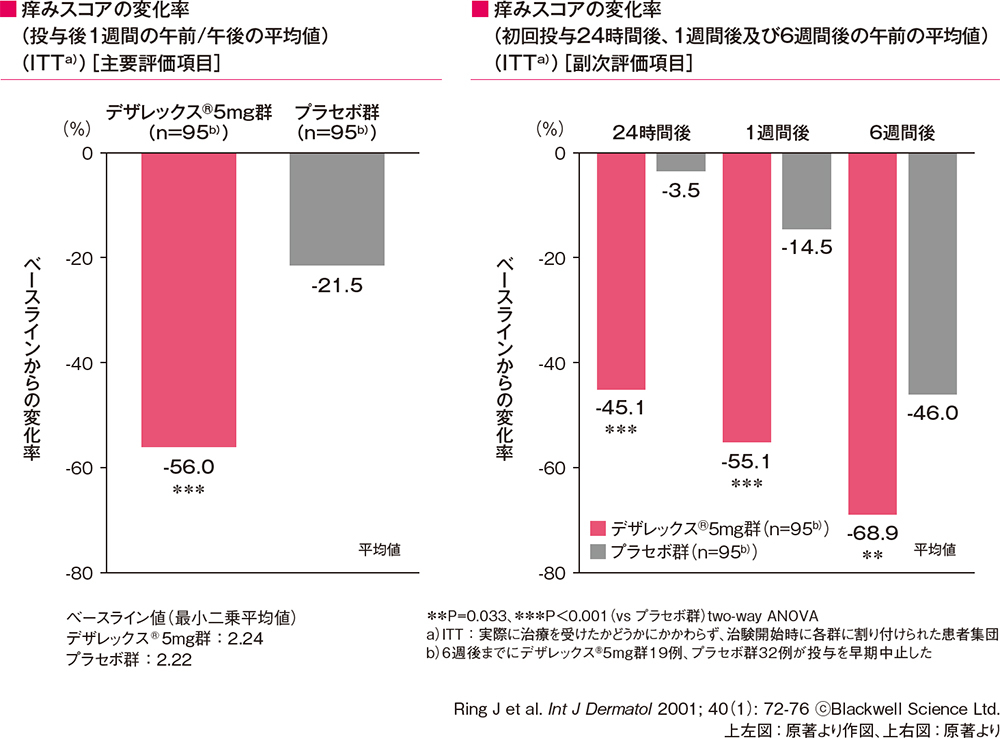
痒みスコアの変化率(投与後1週間の午前/午後の平均値)[主要評価項目]
痒みスコアの変化率(初回投与24時間後、1週間後及び6週間後の午前の平均値)[副次評価項目]
痒みスコアの変化率(投与後1週間の午前/午後の平均値、初回投与24時間後、1週間後及び6週間後の午前の平均値)は、 いずれもデザレックス®5mg群がプラセボ群に比べて有意な低下を示しました(**P=0.033,***P<0.001、two-way ANOVA)。
痒みスコアの変化率
安全性
有害事象の発現頻度は、デザレックス®5mg群が55.8%(53/95例)、プラセボ群が43.2%(41/95例)でした。デザレックス®5mg群で最も発現頻度が高かった有害事象は頭痛(12.6%)であり、次いで疲労(8.4%)、ウイルス感染(7.4%)、咽頭炎(6.3%)、上気道感染(5.3%)及びめまい(5.3%)でした。一方、プラセボ群で最も発現頻度が高かった有害事象は頭痛(16.8%)であり、次いでウイルス感染(8.4%)、上気道感染(4.2%)、咽頭炎(3.2%)、及びめまい(2.1%)でした。また投与中止に至った有害事象は、デザレックス®5mg群で3例、プラセボ群で2例でした。
バイタルサイン、臨床検査パラメータ、心電図基準のベースラインからの臨床的に問題となる変化は、デザレックス®5mg群、プラセボ群で認められませんでした。
- 1)承認時評価資料(蕁麻疹患者対象第Ⅲ相臨床試験)
- 2)Ring J et al. Int J Dermatol 2001; 40(1): 72-76
利益相反:本研究は旧Schering-Ploughの支援により行われた
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。