

-臨床成績〈小児〉比較試験-
国内第Ⅲ相非盲験クロスオーバー比較試験〈小児〉1)
| 目的 |
フルティフォーム®群のフルチカゾン/サルメテロール配合剤(SFC)群に対する非劣性を検証する。またフルティフォーム®群の有効性及び安全性をSFC群と比較する。 |
|---|---|
| 対象 |
5歳以上16歳未満の小児気管支喘息患者87例 |
| 方法 |
多施設共同、実薬対照、無作為化、非盲検、2群2期クロスオーバー比較試験。フルティフォーム®50/5μg又はフルチカゾン/サルメテロール配合剤50/25μgエアゾールをそれぞれ1回2吸入、1日2回、各治療期2週間投与した。投与順1はフルティフォーム®群→SFC群、投与順2はSFC群→フルティフォーム®群とし、年齢を割付因子とした動的割付けを行った。観察期及び休薬期は、観察期前のICS用量を目安にFP(フルチカゾン)エアゾールを100μg/日又は200μg/日投与した。治療期1開始日に、治療期1移行基準を満たした被験者を投与順1又は投与順2に1:1の比で無作為に割り当てた。 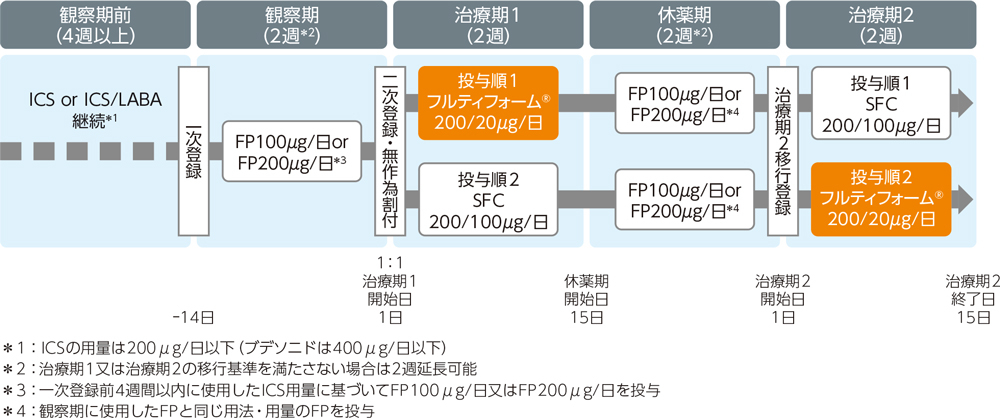
|
| 評価項目 |
有効性評価項目
安全性評価項目 |
| 統計解析 |
主要評価項目
副次評価項目/その他の有効性評価項目 *ピークフロー値は来院前7日間の平均値で、ベースラインは治療期1開始日直前7日間の平均値を用いた。 |
| スペーサーの使用 |
被験者が治験薬及びフルチカゾンプロピオン酸エステル エアゾールを吸入する際には、スペーサーの使用を必須とした。また、本治験で使用するスペーサーは、エアロチャンバー(トゥルーデルメディカル社製)とし、マウスピースタイプ、又はマスクタイプのいずれかを治験責任医師等の判断で選択した。なお、サルブタモール吸入時にはスペーサーの使用を必須としないが、可能な限り使用の有無を統一させた。 |
臨床成績
主要評価項目
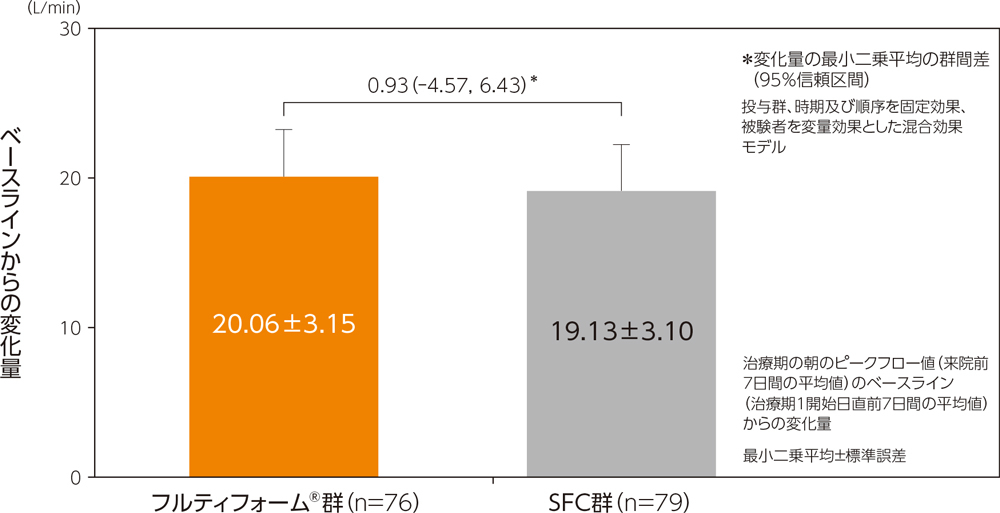
(1)朝のピークフロー値
主要評価項目である朝のピークフロー値のベースラインからの変化量の最小二乗平均は、フルティフォーム®群で20.06L/min、SFC群で19.13L/minであった。フルティフォーム®群のSFC群に対する変化量の最小二乗平均の群間差は、0.93L/min(95%信頼区間:−4.57~6.43L/min)であり、群間差の両側95%信頼区間の下限が−15L/min以上であったことから、SFCに対するフルティフォーム®の非劣性が検証された。
副次評価項目
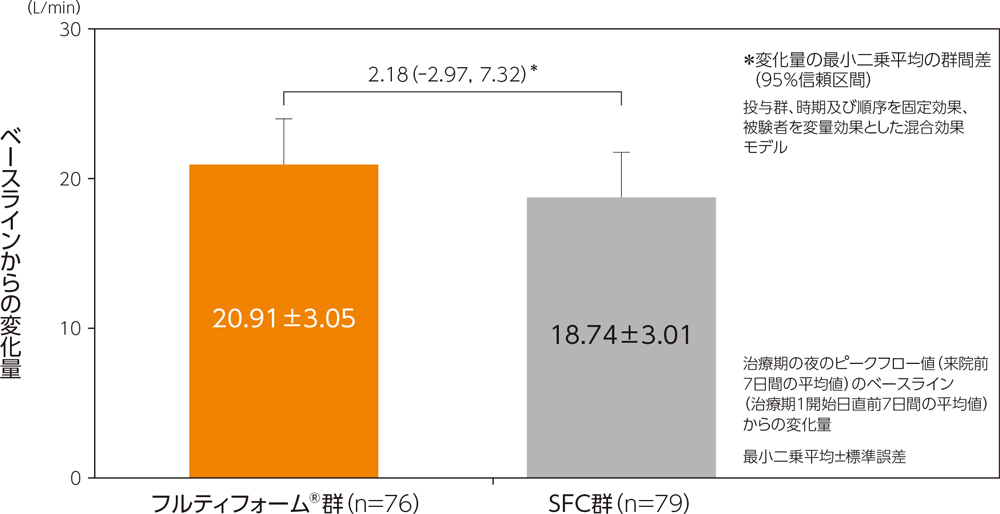
(2)夜のピークフロー値
副次評価項目である夜のピークフロー値のベースラインからの変化量の最小二乗平均は、フルティフォーム®群で20.91L/min、SFC群で18.74L/minであり、最小二乗平均の群間差は、2.18L/min(95%信頼区間:−2.97~7.32L/min)であった。
その他の有効性評価項目
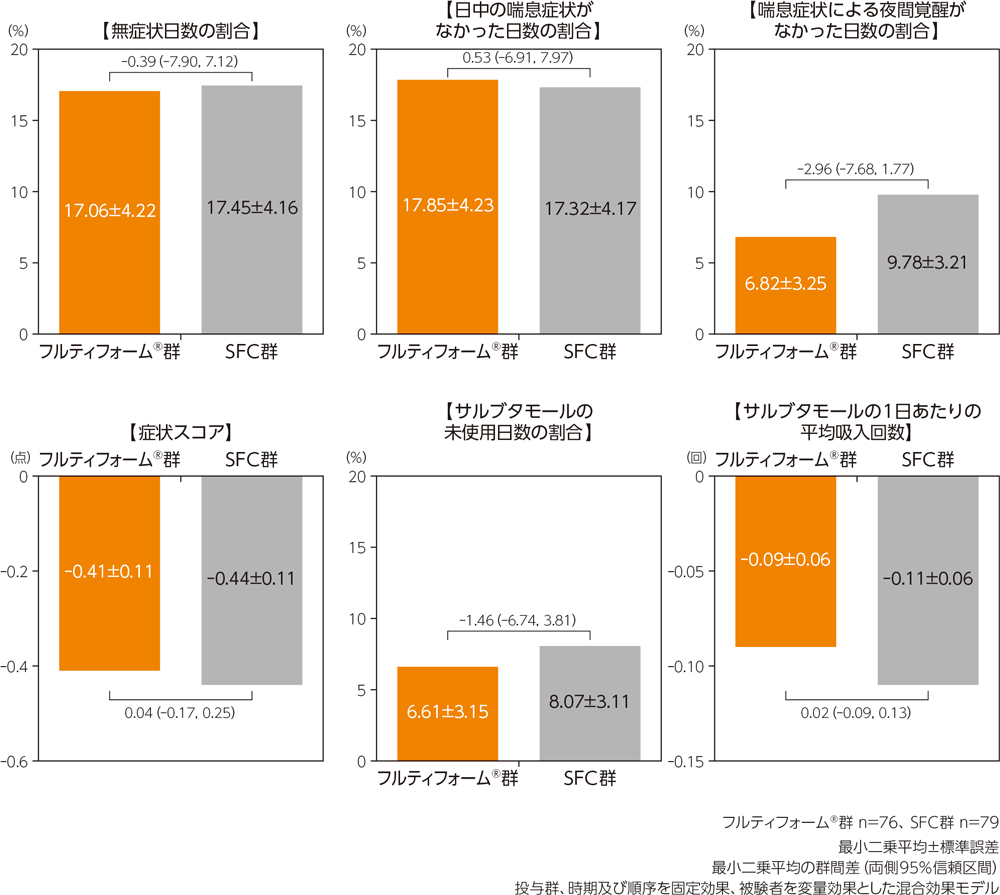
(3)各指標のベースラインからの変化量
治療期におけるベースラインからの変化量の最小二乗平均の群間差は、無症状日数の割合が−0.39%(95%信頼区間:−7.90%~7.12%)、日中の喘息症状がなかった日数の割合が0.53%(95%信頼区間:−6.91%~7.97%)、喘息症状による夜間覚醒がなかった日数の割合が−2.96%(95%信頼区間:−7.68%~1.77%)、症状スコアが0.04点(95%信頼区間:−0.17点~0.25点)、サルブタモールの未使用日数の割合が−1.46%(95%信頼区間:−6.74%~3.81%)、サルブタモールの1日あたりの平均吸入回数が0.02回(95%信頼区間:−0.09回~0.13回)であった。
5. 効能又は効果に関連する注意
- 5.1
-
患者、保護者又はそれに代わる適切な者に対し次の注意を与えること。
本剤は発現した発作を速やかに軽減する薬剤ではないので、急性の発作に対しては使用しないこと。
- 5.2
- 本剤の投与開始前には、患者の喘息症状を比較的安定な状態にしておくこと。特に、喘息発作重積状態又は喘息の急激な悪化状態のときには原則として本剤は使用しないこと。
7. 用法及び用量に関連する注意
症状の緩解がみられた場合は、治療上必要最小限の用量を投与し、必要に応じ吸入ステロイド剤への切り替えも考慮すること。
安全性評価項目
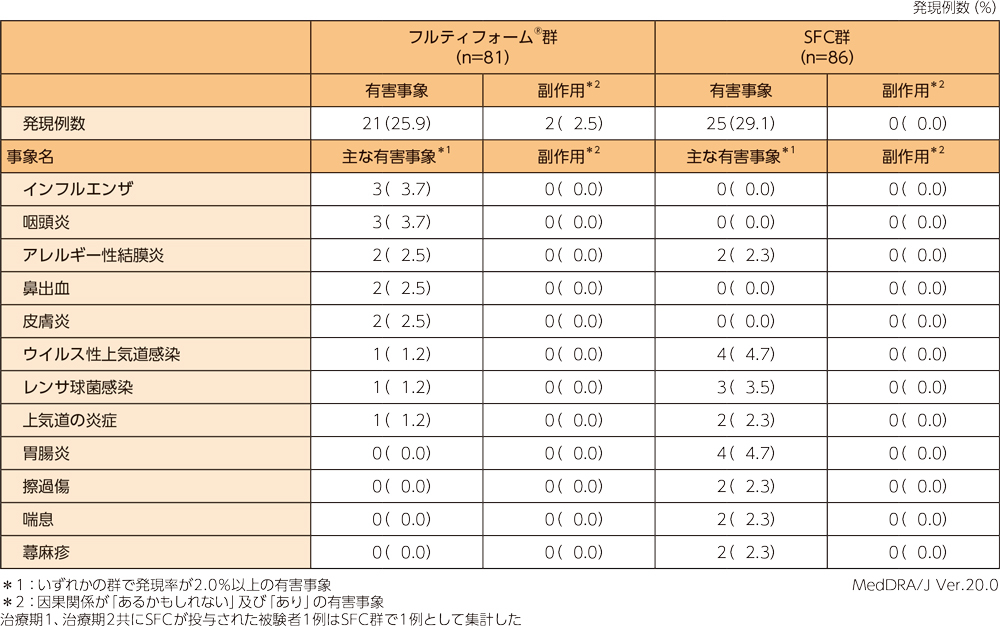
有害事象及び副作用
フルティフォーム®群の有害事象は81例中21例(25.9%)、副作用は81例中2例(2.5%)であった。フルティフォーム®群の主な有害事象は以下のとおりであり、副作用の内訳は頭痛、振戦が各1例であった。本試験において、投与中止に至った有害事象、重篤な有害事象及び死亡例は認められなかった。
9.7 小児等
- 9.7.1.長期間投与する場合には、身長等の経過の観察を十分行うこと。また使用にあたっては、使用法を正しく指導すること。全身ステロイド剤と比較し可能性は低いが、吸入ステロイド剤を特に長期間、大量に投与する場合に成長遅延をきたすおそれがある。なお、小児等に対しては国内での24週間を超える臨床試験は実施していない。
- 9.7.2.低出生体重児、新生児、乳児又は5歳未満の幼児を対象とした臨床試験は実施していない。
- 1)フルティフォーム®の国内第Ⅲ相非盲検クロスオーバー比較試験〈小児〉(承認時評価資料).
禁忌を含む注意事項等情報につきましては電子添文をご参照ください。