

-臨床成績〈小児〉-
臨床成績 -小児気管支喘息に対する国内第Ⅲ相長期投与試験-
フルティフォーム®国内第Ⅲ相長期投与試験〈小児〉(承認時評価資料)
| 目的 |
日本人小児気管支喘息患者を対象として、フルティフォーム® 50/ 5μgを1回2吸入、1日2回、24週間投与したときの安全性及び有効性を検討する。 |
|---|---|
| 対象 |
5歳以上16歳未満の日本人小児気管支喘息患者53例 |
| 方法 |
多施設共同非盲検非対照試験。吸入ステロイド薬(ICS)を4週間以上使用した患者について、観察期として観察期開始前と同じICSを同用量・用法・剤形で2週間継続使用した。治療期移行基準を満たした被験者は、治療期としてフルティフォーム®50/5μgを1回2吸入、1日2回、朝・夜吸入した。治療期移行基準を満たさない被験者は2週間の観察期延長を可能とした。治療期実施期間は24週間とした。 |
| 評価項目 |
|
| 統計解析 |
|
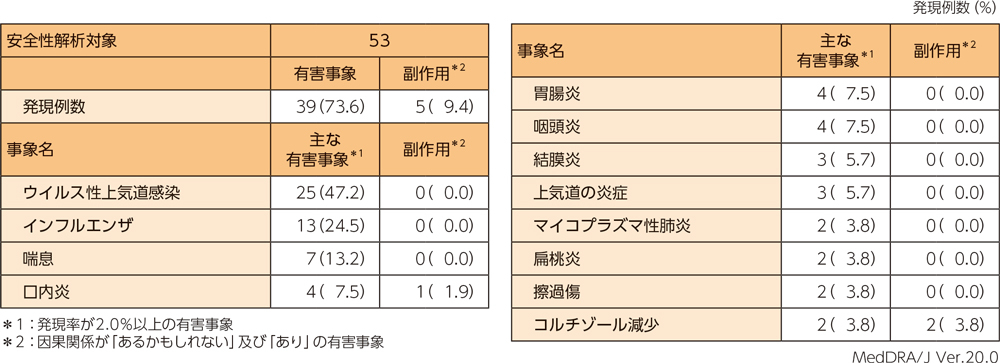
安全性評価項目〈有害事象及び副作用〉
有害事象は53例中39例(73.6%)に認められ、主な有害事象は下表のとおりであった。本試験において重篤な有害事象及び投与中止に至った有害事象、死亡例は認められなかった。副作用は53例中5例(9.4%)に認められ、内訳はコルチゾール減少2例(3.8%)、口内炎及び口腔咽頭不快感、尿中蛋白陽性が各1例(1.9%)であった。
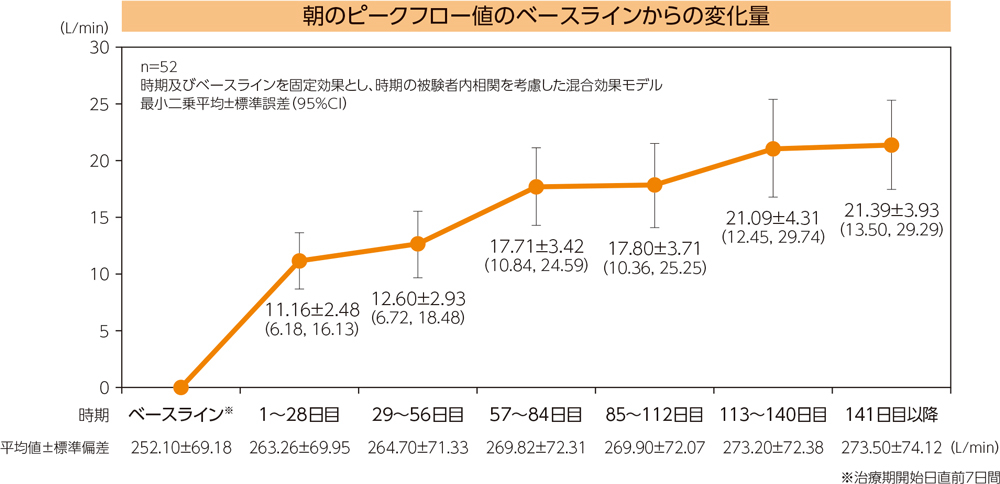
有効性評価項目〈朝のピークフロー値〉
朝のピークフロー値は図のように推移し、最終評価時の変化量は最小二乗平均21.39L/minであった。
9.特定の背景を有する患者に関する注意(抜粋)
9.7 小児等
- 9.7.1
-
長期間投与する場合には、身長等の経過の観察を十分行うこと。また使用にあたっては、使用法を正しく指導すること。
全身ステロイド剤と比較し可能性は低いが、吸入ステロイド剤を特に長期間、大量に投与する場合に成長遅延をきたすおそれがある。なお、小児等に対しては国内での24週間を超える臨床試験は実施していない。
- 9.7.2
- 低出生体重児、新生児、乳児又は5歳未満の幼児を対象とした臨床試験は実施していない。
臨床成績 -小児気管支喘息に対する国内第Ⅲ相非盲検クロスオーバー比較試験-
フルティフォーム®国内第Ⅲ相非盲検クロスオーバー比較試験〈小児〉(承認時評価資料)
| 目的 |
フルティフォーム® 群のSFC(フルチカゾン/サルメテロール配合剤)群に対する非劣性を検証する。またフルティフォーム®群の有効性及び安全性をSFC群と比較する。 |
|---|---|
| 対象 |
5歳以上16歳未満の小児気管支喘息患者87例 有効性解析対象集団(PPS) フルティフォーム®群76例 SFC群79例 |
| 方法 |
多施設共同無作為化非盲検2群2期クロスオーバー比較試験。フルティフォーム®50/5μg又はSFC50/25μgエアゾールをそれぞれ1回2吸入、1日2回、各治療期2週間投与した。投与順1はフルティフォーム®群→SFC群、投与順2はSFC群→フルティフォーム®群とし、年齢を割付因子とした動的割付けを行った。観察期及び休薬期はFP(フルチカゾン)エアゾールを100μg/日又は200μg/日投与した。 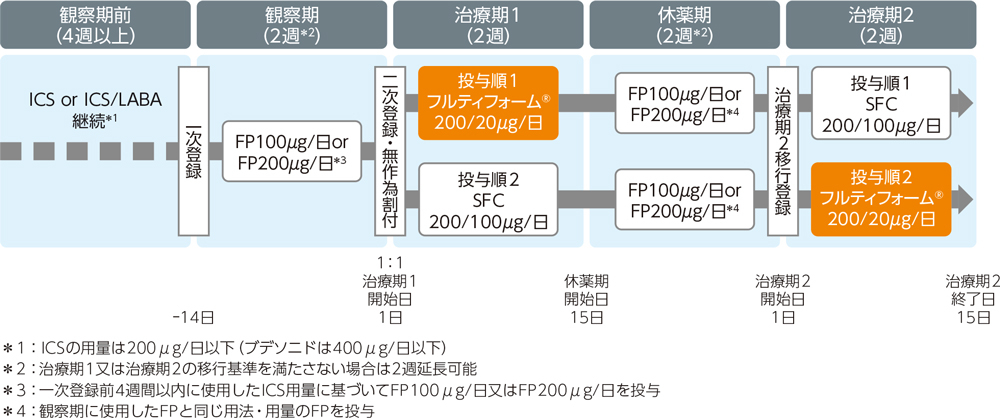
|
| 評価項目 |
|
| 解析計画 |
|
スペーサーの使用
被験者が治験薬及びフルチカゾンプロピオン酸エステル エアゾールを吸入する際には、スペーサーの使用を必須とした。また、本治験で使用するスペーサーは、エアロチャンバー(トゥルーデルメディカル社製)とし、マウスピースタイプ、又はマスクタイプのいずれかを治験責任医師等の判断で選択した。なお、サルブタモール吸入時にはスペーサーの使用を必須としないが、可能な限り使用の有無を統一させた。
有効性
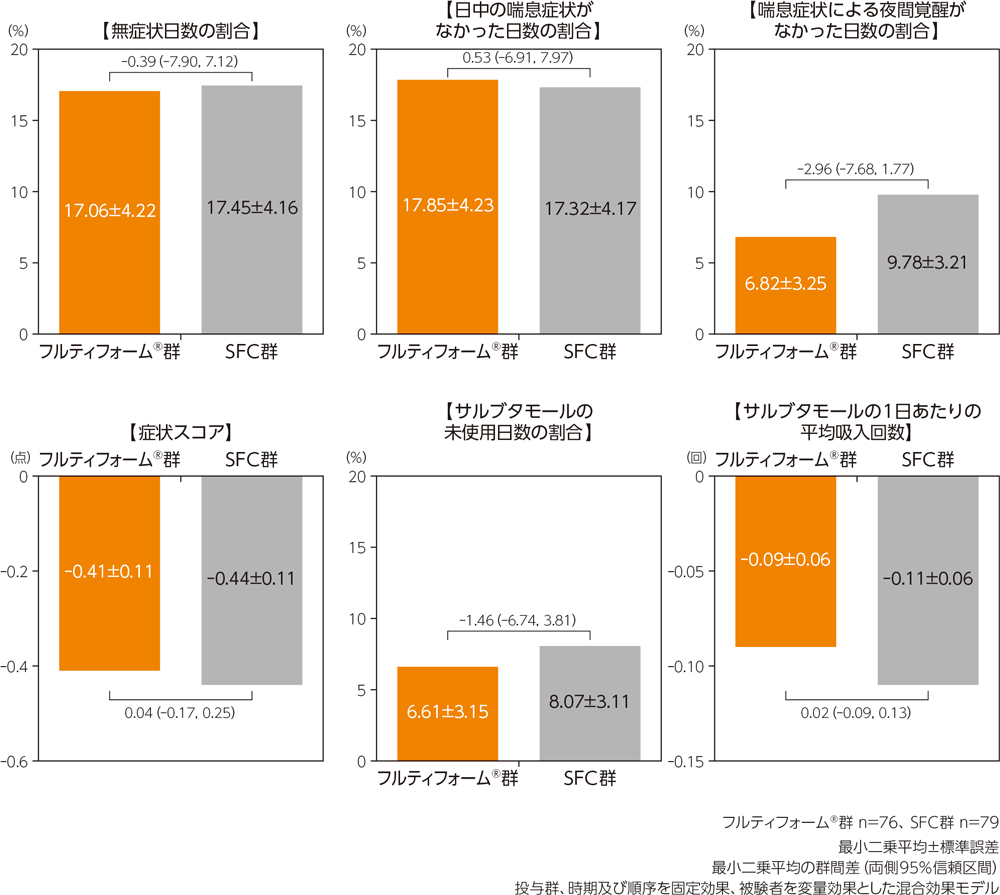
朝のピークフロー値のベースラインからの変化量について、最小二乗平均の群間差(95%信頼区間)は0.93(-4.57,6.43)であり、両側95%信頼区間の下限が事前規定した非劣性の限界値(-15L/min)を上回ったため、SFCに対するフルティフォーム®の非劣性が検証された。
最小二乗平均±標準誤差、最小二乗平均の差(95%信頼区間)
投与群、時期及び順序を固定効果、被験者を変量効果とした混合効果モデル
※非劣性限界値:-15L/min
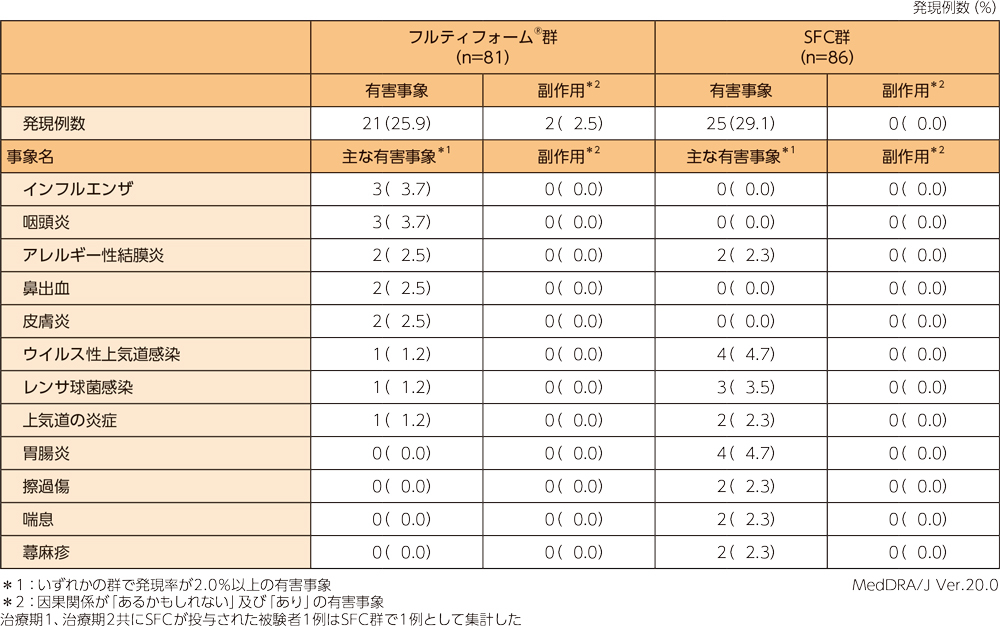
安全性〈有害事象及び副作用〉
有害事象は、フルティフォーム®群では81例中21例(25.9%)、SFC群では86例中25例(29.1%)に認められ、主な有害事象は下表のとおりであった。両群ともに、本試験において重篤な有害事象及び投与中止に至った有害事象、死亡例は認められなかった。
副作用は、フルティフォーム®群では81例中2例(2.5%)に認められ、内訳は頭痛、振戦が各1例(1.2%)であった。
SFC群では認められなかった。
SFC:フルチカゾン/サルメテロール配合剤